SEED | mRNA 2.0:从疫苗走向药物 SEED | mRNA 2.0 beyond vaccines AI-assisted · reviewed
Karolinska Institutet 的 Kenneth R. Chien、Kylie S. Foo 与 Nevin Witman 团队近期撰写的 Review 梳理了 mRNA 技术从疫苗走向治疗性药物时遇到的临床证据、递送瓶颈和平台分化,为理解 mRNA therapeutics 2.0 提供了一张更清晰的转化地图。
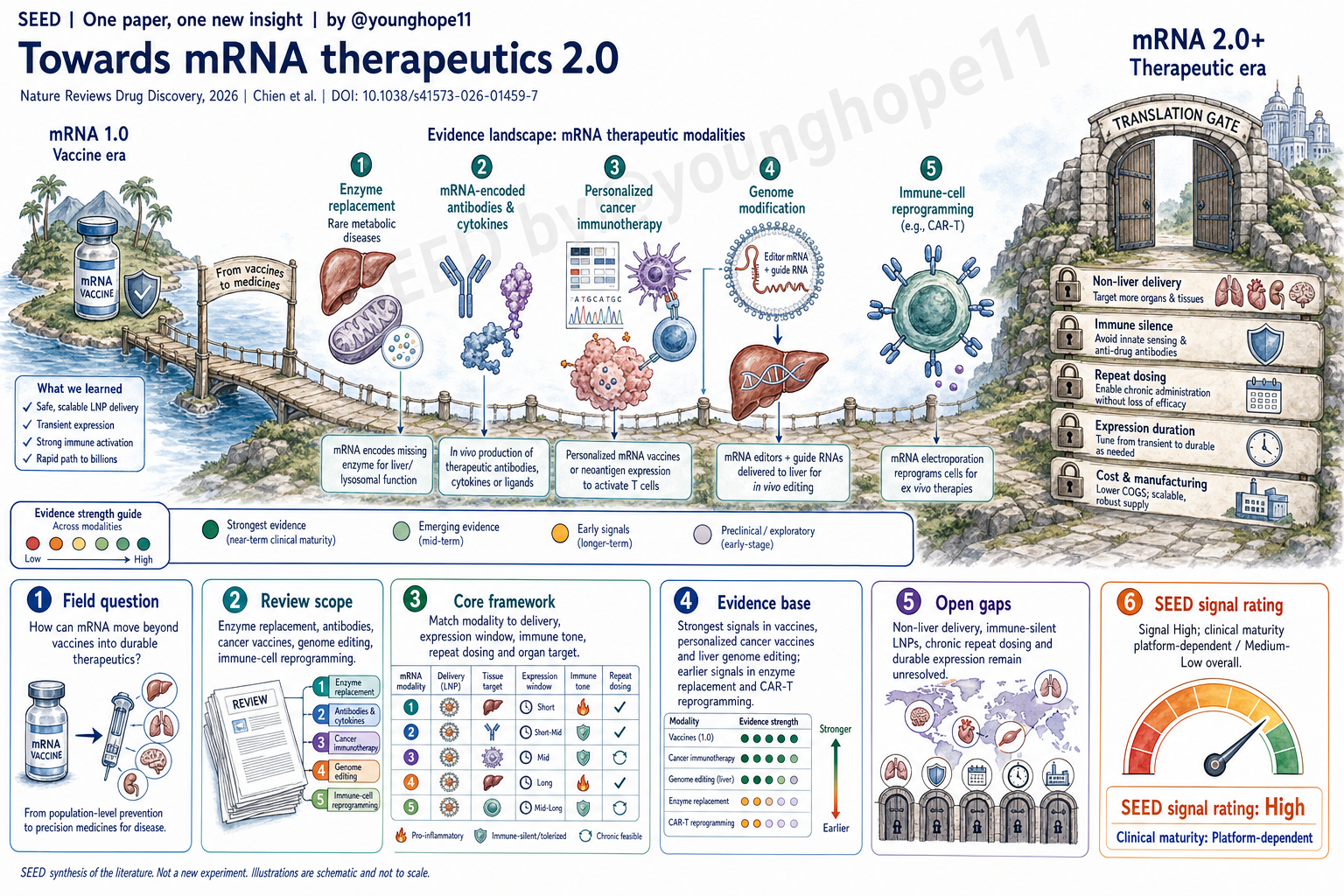
这篇 Review 试图回答什么问题?
这篇 Review 问的是:mRNA 已经证明可以快速制造、规模化生产并诱导强免疫反应,但它能否真正从疫苗平台变成一类可反复、可控、可器官定向使用的治疗性药物?
疫苗场景里,短暂表达和免疫刺激常常是优点。治疗场景则不同。酶替代、抗体表达、细胞因子调控、个体化癌症免疫治疗、体内基因编辑和免疫细胞重编程,对递送器官、表达时长、剂量窗口、免疫反应和重复给药的要求完全不同。
因此,文章真正整理的不是“mRNA 能做什么”的愿望清单,而是疫苗之后的 mRNA 药物化问题:哪些方向已经有临床信号,哪些仍主要是平台承诺,以及 mRNA 2.0 还缺哪些工程能力。
它真正整理出的新框架是什么?
它整理出的框架可以概括为一句话:mRNA therapeutics 2.0 不是单一平台升级,而是把不同治疗模式匹配到不同的递送、表达和免疫约束。
对于 secreted protein 或 paracrine factor,短暂局部表达可能就足够,例如 VEGFA mRNA 用来诱导局部再生性血管生成。对于 enzyme replacement,问题立刻变成长期或重复给药,因为患者需要持续补充缺失蛋白。对于 mRNA-encoded antibodies 和 cytokines,肝脏可以暂时成为体内蛋白工厂,但系统性暴露、炎症和药代动力学需要被精细控制。
对于 personalized cancer immunotherapy,mRNA 的优势是速度:肿瘤测序、neoantigen 选择、mRNA 制备和给药必须在临床时间窗内完成。对于 genome editing,mRNA 的短暂表达反而是优点,因为 Cas9、base editor 或 prime editor 不应长期存在。对于 immune-cell reprogramming,mRNA 可以让 CAR 表达更可逆,也可能把 ex vivo 细胞治疗推向更可规模化的 in vivo 重编程。
这套框架最后收束到几个共同瓶颈:clinically tractable in vivo delivery、对肝脏以外器官的偏向性递送、足够 immune-silent 的 mRNA-vehicle formulation、可重复给药、合适的表达时长,以及成本和制造可放大性。
证据链强在哪里?
强在文章没有只讨论化学修饰或 LNP 配方,而是把十多年临床探索放进同一张转化地图里。
第一条证据线是 secreted protein therapeutics。VEGFA mRNA 是疫苗以外较早进入人体试验的 mRNA 治疗思路,后续 AZD8601 在小型 phase II 心衰研究中没有因为样本量太小而证明明确疗效,但显示了局部 mRNA 给药、短暂蛋白表达和安全性探索的可行性。这条线说明,局部或旁分泌蛋白可能是 mRNA 药物较自然的入口,但全器官递送和持久表达仍然困难。
第二条证据线是 rare disease enzyme replacement。propionic acidaemia 的 LNP-mRNA 方案同时编码 PCCA 和 PCCB 两个亚基,在 16 名患者的中期分析中,让有既往 metabolic decompensation events 的 8 名患者风险下降约 70%。这很有启发性,因为它证明 mRNA 可以在体内表达多亚基、亚细胞定位相关的酶;但同一案例也提醒我们,显著 treatment-related adverse events 和慢性给药频率仍是关键问题。
第三条证据线是 personalized mRNA cancer immunotherapy。mRNA-4157/intismeran autogene 联合 pembrolizumab 在高风险切除后 melanoma 的 phase IIb 研究中显示复发风险下降,开发方公布 5 年随访风险降低 49%,phase III 仍在进行。PDAC 中 autogene cevumeran 的 phase I 结果也显示,部分患者出现长期功能性 T 细胞反应,并与复发延迟相关。这条线说明,mRNA 在“快速、个体化、多抗原”场景中有真实临床吸引力。
第四条证据线是 genome modification。NTLA-2001 把 Cas9 mRNA 和 guide RNA 通过 LNP 递送到肝脏,用于 ATTR 的体内基因破坏,并已经进入 phase III。PCSK9 base editing 则展示了“one and done”治疗常见病风险因子的想象力,同时 VERVE-101 的安全信号和后续 VERVE-102、GalNAc-LNP 路线也说明:编辑器和递送载体都是新技术时,安全窗口会成为核心考题。
第五条证据线是 immune-cell reprogramming。Descartes-08 这类 mRNA CAR-T 方案在 generalized myasthenia gravis 的 phase IIb 中报告了 12 个月内的持续反应和可接受安全性,phase III 已启动;而 in vivo CAR-T 重编程仍主要处在 preclinical 阶段。这条线展示了 mRNA 连接细胞治疗和可逆表达的潜力。
最大局限是什么?
最大局限是这是一篇叙述性 Review,而不是 systematic review 或 meta-analysis。它选择了一组代表性临床研究来总结 mRNA 药物化路线,但不同项目的疾病、剂量、给药方式、payload、终点和临床阶段差异很大,不能被读成严格的头对头比较。
第二个局限是很多证据仍然早期、开放标签、小样本,或者来自公司公告和中期结果。melanoma 个体化疫苗、ATTR 编辑、propionic acidaemia 酶替代和 mRNA CAR-T 都很重要,但它们分别处在不同成熟度上。对大多数非疫苗 mRNA therapeutics 来说,临床疗效、长期安全、重复给药和真实世界可制造性仍需要更长时间验证。
第三个局限是文章所强调的核心瓶颈本身还没有解决。肝脏递送已经相对成熟,但许多肝脏靶点也可以被 siRNA 更简单地处理。真正可能打开新空间的是心脏、骨骼肌、肾脏、肺、造血系统和特定免疫细胞等非肝组织递送;这些方向仍缺少足够稳定、可放大、可监管的临床证明。
最后,mRNA 的优势和风险在不同应用中会反转。免疫刺激对疫苗是优势,对慢性治疗可能是毒性;短暂表达对 genome editing 是安全边界,对 enzyme replacement 却是给药负担。这也是为什么不能用“mRNA 平台成熟”来概括所有 mRNA 药物。
对转化或领域判断有什么意义?
这篇 Review 的价值在于帮助判断哪些 mRNA 方向更接近药物化,哪些还停留在平台早期。
我会把 mRNA therapeutics 2.0 拆成三层。第一层是已经有强平台经验的疫苗和部分肝脏递送场景,这里 mRNA 和 LNP 的制造、监管和人体经验较多。第二层是正在形成临床信号的 personalized cancer vaccines、肝脏 genome editing、部分 rare disease enzyme replacement 和 mRNA CAR-T,这些方向已经不是纯概念,但仍要经受 phase III、长期安全和规模化制造检验。第三层是更具想象力的非肝递送、in vivo immune-cell reprogramming、oral or inhaled mRNA、circRNA/self-amplifying RNA 和 AI-guided formulation,这些可能决定 mRNA 2.0 的上限,但临床成熟度还低。
对研发决策来说,这篇文章给出的判断标准很直接:先问 therapeutic modality 需要短暂表达还是持久表达;需要局部、肝脏还是非肝递送;免疫反应是需要被激活还是被压低;能不能重复给药;剂量是否足够低;制造和成本是否能支持目标适应症。
它也提醒我们,mRNA 2.0 的突破点可能不只是更好的 mRNA 序列,而是 delivery technology。只有当递送能够安全、剂量依赖、细胞类型特异地进入目标组织时,mRNA 才能从“疫苗时代的平台”变成更广泛的 therapeutic operating system。
Yang 的信号评级:High
理由:我会把这篇 Review 的科研信号评为 High。它不是因为提出了一个新实验结果而 High,而是因为它把疫苗之后的 mRNA 药物化问题整理成了一个清楚的转化框架:不同应用需要不同的递送、表达窗口、免疫状态、重复给药能力和制造路径。
这个 High 是“领域判断信号 High”,不是“临床就绪 High”。文章最强的地方在于它帮助读者区分三件事:mRNA 已经证明的能力、早期临床正在验证的能力,以及 mRNA 2.0 仍必须补上的工程能力。
临床成熟度我会单独评为 platform-dependent,整体大约是 Medium-Low。疫苗成熟度很高,肝脏 LNP 和部分癌症疫苗/编辑项目正在接近更强临床验证;但慢性 enzyme replacement、非肝递送、immune-silent repeat dosing 和 in vivo immune-cell reprogramming 仍然需要更多人体数据。
Kenneth R. Chien, Kylie S. Foo and Nevin Witman at Karolinska Institutet recently wrote a Review that organizes the clinical evidence, delivery bottlenecks and platform branching required for mRNA technology to move from vaccines into therapeutic medicines, providing a clearer translation map for mRNA therapeutics 2.0.
What question does this review try to answer?
This review asks whether mRNA, after proving that it can be rapidly designed, manufactured at scale and used to induce strong immune responses, can become a repeatable, controllable and organ-targeted therapeutic drug class.
The vaccine setting is unusual: transient expression and immune stimulation are often useful. Therapeutic settings are different. Enzyme replacement, antibody expression, cytokine modulation, personalized cancer immunotherapy, in vivo genome editing and immune-cell reprogramming each impose different requirements on delivery organ, expression duration, dose window, immune response and repeat dosing.
The article is therefore not a wish list of everything mRNA could do. It is a map of post-vaccine mRNA drug development: which directions already have clinical signals, which remain mostly platform promises, and what engineering capabilities mRNA 2.0 still lacks.
What framework does it synthesize?
The framework can be summarized in one sentence: mRNA therapeutics 2.0 is not a single platform upgrade, but a matching problem between therapeutic modality and delivery, expression and immune constraints.
For secreted proteins or paracrine factors, transient local expression may be sufficient, as illustrated by VEGFA mRNA for regenerative angiogenesis. For enzyme replacement, the problem quickly becomes durable or repeated dosing, because patients need sustained replacement of a missing protein. For mRNA-encoded antibodies and cytokines, the liver can temporarily act as an in vivo protein factory, but systemic exposure, inflammation and pharmacokinetics have to be tightly controlled.
For personalized cancer immunotherapy, the advantage of mRNA is speed: tumor sequencing, neoantigen selection, mRNA manufacturing and treatment must fit inside a clinical time window. For genome editing, transient mRNA expression becomes a safety feature, because Cas9, base editors or prime editors should not persist indefinitely. For immune-cell reprogramming, mRNA can make CAR expression more reversible and may eventually push ex vivo cell therapy toward more scalable in vivo reprogramming.
The framework converges on a shared set of bottlenecks: clinically tractable in vivo delivery, preferential delivery beyond the liver, sufficiently immune-silent mRNA-vehicle formulations, repeat dosing, fit-for-purpose expression duration, and scalable cost and manufacturing.
Where is the evidence base strongest?
The evidence base is strongest because the review does not focus only on chemical modification or LNP formulation. It places more than a decade of clinical exploration onto one translation map.
The first evidence line is secreted protein therapeutics. VEGFA mRNA was among the earliest non-vaccine mRNA therapeutic concepts tested in humans. The later AZD8601 phase II heart-failure study was too small to establish clear efficacy, but it explored local mRNA delivery, transient protein expression and safety. This line suggests that local or paracrine proteins may be a natural entry point for mRNA medicines, while whole-organ delivery and durable expression remain difficult.
The second evidence line is rare-disease enzyme replacement. In propionic acidaemia, an LNP-mRNA therapy encoding both PCCA and PCCB subunits reduced the risk of metabolic decompensation events by about 70% among the eight participants who had such events during the pretreatment period in an interim analysis of 16 patients. This is instructive because it shows that mRNA can express multi-subunit, subcellularly localized enzymes in vivo. The same example also highlights the importance of treatment-related adverse events and chronic dosing frequency.
The third evidence line is personalized mRNA cancer immunotherapy. mRNA-4157, or intismeran autogene, combined with pembrolizumab reduced recurrence risk in high-risk resected melanoma in a phase IIb trial; the developers reported a 49% risk reduction after five years of follow-up, while phase III testing continues. In PDAC, phase I results with autogene cevumeran showed long-term functional T cell responses in some patients and association with delayed recurrence. This line shows that mRNA has real clinical pull where speed, personalization and multiplexed antigens matter.
The fourth evidence line is genome modification. NTLA-2001 uses LNP delivery of Cas9 mRNA and guide RNA to disrupt TTR in the liver for ATTR, and has moved into phase III trials. PCSK9 base editing shows the appeal of a one-and-done approach for a common disease risk factor, while the safety signal with VERVE-101 and the move toward VERVE-102 and GalNAc-LNP designs show that the safety window becomes the central test when both editor and delivery vehicle are new technologies.
The fifth evidence line is immune-cell reprogramming. mRNA CAR-T approaches such as Descartes-08 have reported durable responses and an acceptable safety profile over 12 months in a phase IIb generalized myasthenia gravis trial, with phase III underway. In vivo CAR-T reprogramming remains mainly preclinical, but it shows how mRNA could connect cell therapy with reversible expression and greater scalability.
What is the biggest limitation?
The biggest limitation is that this is a narrative Review, not a systematic review or meta-analysis. It selects a representative group of clinical studies to organize the mRNA therapeutic landscape, but the projects differ in disease, dose, administration route, payload, endpoint and clinical stage. It should not be read as a strict head-to-head comparison.
A second limitation is that much of the evidence remains early, open-label, small, interim or company-reported. The melanoma personalized vaccine, ATTR editing, propionic acidaemia enzyme replacement and mRNA CAR-T examples are all important, but they sit at different levels of maturity. For most non-vaccine mRNA therapeutics, clinical efficacy, long-term safety, repeat dosing and real-world manufacturability still need longer validation.
A third limitation is that the core bottlenecks emphasized by the article are still unsolved. Liver delivery is relatively mature, but many liver targets can also be addressed more simply with siRNAs. The larger opportunity may lie in delivery to heart, skeletal muscle, kidney, lung, haematopoietic lineages and specific immune cells. Those areas still lack stable, scalable and regulator-ready clinical proof.
Finally, mRNA’s strengths and risks reverse across applications. Immune stimulation is an advantage for vaccines but can become toxicity for chronic therapy. Transient expression is a safety boundary for genome editing but a dosing burden for enzyme replacement. That is why it is misleading to describe all mRNA medicines as clinically mature just because the platform has succeeded in vaccines.
What does it mean for translation or field-level judgment?
The value of this Review is that it helps judge which mRNA directions are closer to real drug development and which remain earlier platform bets.
I would divide mRNA therapeutics 2.0 into three layers. The first layer includes vaccines and some liver-delivery settings, where mRNA and LNP manufacturing, regulatory and human experience are strongest. The second layer includes directions with emerging clinical signal: personalized cancer vaccines, liver genome editing, some rare-disease enzyme replacement and mRNA CAR-T. These are no longer pure concepts, but still need phase III validation, long-term safety and scalable manufacturing. The third layer includes more expansive opportunities: non-liver delivery, in vivo immune-cell reprogramming, oral or inhaled mRNA, circRNA or self-amplifying RNA, and AI-guided formulation. These may define the upper limit of mRNA 2.0, but their clinical maturity remains low.
For R&D decisions, the review’s decision criteria are direct. Ask first whether the therapeutic modality needs transient or durable expression; local, liver or non-liver delivery; immune activation or immune silence; repeat dosing; a sufficiently low dose; and manufacturing economics that fit the intended indication.
It also makes clear that the decisive mRNA 2.0 breakthrough may not be only a better mRNA sequence. It may be delivery technology. Only when delivery becomes safe, dose-dependent and cell-type-specific in target tissues can mRNA move from a vaccine-era platform into a broader therapeutic operating system.
Yang’s signal rating: High
Reason: I would rate the scientific signal of this Review as High. The High does not come from one new experimental result. It comes from the way the article turns post-vaccine mRNA drug development into a clear translation framework: different applications require different delivery systems, expression windows, immune states, repeat-dosing capabilities and manufacturing paths.
This High means field-judgment signal High, not clinical readiness High. The article is most useful because it helps readers separate three things: what mRNA has already proved, what early clinical studies are now testing, and what engineering capabilities mRNA 2.0 still needs.
I would rate clinical maturity separately as platform-dependent, roughly Medium-Low overall. Vaccine maturity is high, and liver LNP plus some cancer vaccine and editing programs are approaching stronger clinical validation. But chronic enzyme replacement, non-liver delivery, immune-silent repeat dosing and in vivo immune-cell reprogramming still need more human data.