SEED | Shred-seq:把人类基因组切成 essentiality 地图 SEED | Shred-seq maps genome essentiality by deletion AI-assisted · reviewed
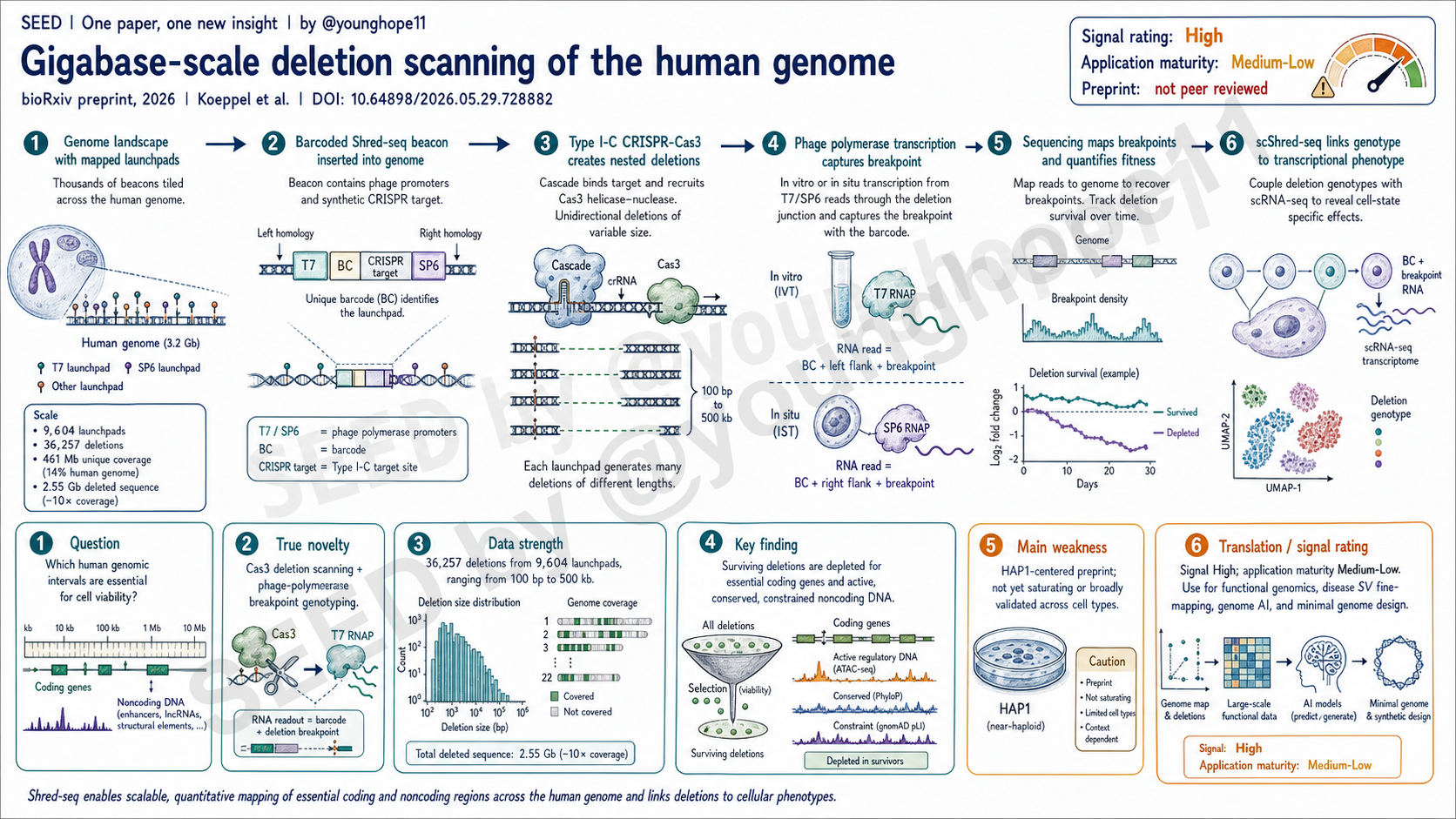
University of Washington、Seattle Hub for Synthetic Biology 和 Brotman Baty Institute 的 Jonas Koeppel 与 Jay Shendure、Sudarshan Pinglay 团队近期在 bioRxiv 发布预印本,提出 Shred-seq,将 CRISPR-Cas3 大片段 deletion 与 phage polymerase-based breakpoint genotyping 结合,为系统性扫描人类基因组中 coding 与 noncoding 区间的细胞 essentiality 提供了新的方法平台。
研究问题是什么?
这篇文章问的是一个 functional genomics 的底层问题:人类基因组中,到底哪些连续 DNA 区间对细胞生存和增殖是必需的?
过去这个问题主要在 ORF 层面被回答。CRISPR knockout 可以系统性识别 protein-coding gene 的 essentiality,但对 noncoding genome、multi-kb regulatory landscapes、large structural variants 和 genome architecture 的需求程度,仍缺少可扩展的实验测量。比较基因组学、自然人群 SV 和 chromatin annotations 都能给出间接线索,但它们不能直接告诉我们:删除某一段连续 DNA 以后,细胞是否还能活、还能增殖、转录状态会怎样。
因此,这篇预印本的核心问题不是某一个基因是否 essential,而是能否建立一种 genome-scale deletion scan,让 coding、noncoding 和大尺度 genomic interval 都能被直接扰动、直接分型、直接连接到 fitness 或单细胞表型。
真正的新意是什么?
真正的新意是 Shred-seq 把两个原本分开的能力接在了一起:Cas3 负责制造大范围、单向、长度可变的 deletion;phage polymerase promoter 负责把不可预测的 breakpoint 变成可测序的分型信号。
具体来说,Shred-seq 先把带有 unique barcode、synthetic CRISPR target 和 T7/SP6 phage promoter 的 beacon 插入基因组。随后 Type I-C CRISPR-Cas3 从 beacon 附近启动,沿 DNA 产生从数百 bp 到数百 kb 的 nested deletions。因为 deletion 终点不可预先知道,传统 PCR primer 或 clonal whole-genome sequencing 很难扩展到全基因组 pooled screen;Shred-seq 的关键设计,是让 phage polymerase 从 beacon promoter 开始转录,直接读出 barcode 和新形成的 genomic junction。
这使 deletion scan 从“一个 locus 一个实验”变成“成千上万个 launchpad 同时读出”。更重要的是,同一套结构可以用 in vitro transcription 做 bulk fitness readout,也可以用 in situ transcription 加 scRNA-seq 做 scShred-seq,把具体 deletion genotype 与单细胞 transcriptome 连接起来。
数据强在哪里?
数据强在它不只是提出概念,而是把方法从 proof-of-concept 推到 gigabase-scale。
在初始 HAP1 proof-of-concept 中,作者用 PiggyBac 整合 Shred-seq beacon,获得 10,744 个 barcode,其中 6,549 个能映射到唯一基因组位置。Cas3 编辑后,GFP-negative 细胞比例达到 34-36%,而对照为 3.4%;T7-IVT breakpoint mapping 在单个实验中识别出 3,603 个 unique deletions,总长度 255 Mb,覆盖 151 Mb 人类参考基因组。deletion 长度呈右偏长尾分布,中位数约 33.1 kb,最大接近 495 kb,约 24% 超过 100 kb。
随后作者优化 beacon 结构。T7 promoter padding 带来 1.7 倍读出提升,而加入 T7/SP6 非对向 promoter 的 v3 beacon 相比 v1 提升 18.3 倍。作者还比较 PiggyBac、lentivirus 和 Sleeping Beauty 的整合偏好,显示不同 delivery modality 会偏向不同 chromatin context,因此可以按实验问题选择 launchpad 分布。
真正的规模来自四个 large-scale Shred-seq screens。合并后,作者恢复了 36,257 个 deletions,来自 9,604 个 Cas3 launchpads,覆盖 461 Mb unique sequence,相当于 13.9% 的人类参考基因组,总 deleted sequence 达 2.55 Gb。在覆盖深度上,3.8% 的参考基因组有至少 5 个 independent deletions,1.8% 有至少 10 个 independent deletions;随机分布模型下,这种高覆盖比例会低约 200 倍。
方法学机制也有实证闭环。Cas3-induced deletion 的长度更符合 log-normal distribution,pre-selection deletion 的拟合中位数 25.8 kb,接近观测中位数 27.5 kb。作者映射了 7,467 个 deletion start sites,发现最强启动峰在 protospacer 末端下游 22-34 nt,约占 25.1%。breakpoint repair 方面,66.5% junction 带 1-10 bp microhomology,提示 MMEJ 是 Cas3 lesion 的主要修复机制,19.2% 带 insertion,14.1% 是 perfect junction。
最关键的生物学读出来自 selection over time。在有 pre/post-selection 时间点的实验中,34,515 个 deletions 覆盖 2.42 Gb cumulative sequence。haploid HAP1 中,post-selection deletions 明显避开 essential protein-coding genes;这种 shift 在 diploid HAP1 中消失,符合第二等位基因缓冲。更重要的是,即使只看 12,049 个不 overlap protein-coding gene 的 noncoding deletions,post-selection deletions 仍然避开 active chromatin、A compartments、早复制、高 GC 和部分功能性/约束性序列,说明 selection 不只是 ORF 效应。
最后,scShred-seq 证明了这个平台能走出 bulk viability readout。作者从 20,000 个 input cells 生成 matched transcriptome 和 T7 IST libraries,QC 后保留 13,944 个 single-cell profiles,其中 9,951 个有较高 T7 IST recovery,4,680 个细胞获得 high-confidence deletion annotations。在 chrX 的一个 gene-dense region,RBM3-overlapping deletions 显著降低 RBM3 表达,RBM3 成为 genome-wide differential expression 最强 hit,说明 deletion genotype 可以被直接连接到转录表型。
最大弱点是什么?
最大弱点是这仍是 bioRxiv 预印本,尚未经过同行评议。文章的数据规模和方法设计很强,但一些关键判断仍需要同行评议、独立实验室复现和更广泛场景验证。
第二个限制是实验系统主要集中在 HAP1,尤其是 haploid 或 near-haploid、cancer-derived cell line。cellular viability 的 essentiality 强烈依赖 cell type、ploidy、growth condition 和 developmental state;HAP1 中“可删除”或“不可删除”的区间,不能直接外推到 primary cells、stem cells、differentiated lineages 或 organismal viability。
第三个限制是 coverage 还不是真正的 genome-wide saturation。461 Mb unique coverage 已经很大,但仍只覆盖约 14% 人类参考基因组。作者给出的 haploid genome dispensable fraction 下界约 50%、上界约 96%,这个范围本身说明当前估计仍很宽,主要价值在于证明方法可行,而不是给出最终的人类最小基因组答案。
第四个限制是平台仍有工程瓶颈。flow sorting 限制规模;随机 integration 会偏向 open/active chromatin,heterochromatin 和 polycomb-repressed DNA 覆盖不足;Cas3 的 deletion length distribution 还不能按需求精确控制;scShred-seq 虽然很有前景,但成本、throughput 和统计功效仍限制细微效应、罕见表型和组合效应检测。
是否有临床转化意义?
它不是临床论文,也不应被解读为直接治疗工具。但它有很强的平台转化意义。
第一,它为 disease-associated structural variants 的功能精细定位提供了新路线。很多人类疾病 SV,例如 recurrent deletion 或 duplication,往往跨越多个 genes、enhancers 和 regulatory elements。Shred-seq 的 targeted 版本可以在这些区域建立 dense nested deletion series,帮助定位真正驱动表型的 sub-sequence。
第二,它可能为 noncoding genome 的功能图谱和 genome AI 提供训练数据。现在很多 perturbation datasets 以 ORF-level Perturb-seq 为主,noncoding interval、large SV 和 genome architecture 的 perturbation-phenotype pairs 很少。Shred-seq 可以生成不同长度、不同 genomic context 的 deletion 与 fitness/transcriptome readout,这类数据对 variant effect prediction、generative genome model 和 virtual cell model 都很有价值。
第三,它为 synthetic biology 和 minimal human genome 提供了实验路径。作者讨论了 top-down progressive deletion 和 bottom-up genome synthesis 的可能性。这个方向现在还很远,但 Shred-seq 提供的是一种能直接测试“哪段 DNA 可删除”的工具,而不是只靠 conservation 或 annotation 推断。
所以,这篇文章的转化意义更接近“功能基因组学基础设施”,不是临床成熟产品。它可能影响疾病 SV 解析、合成基因组设计、细胞工程和 AI 训练数据,但实际应用成熟度仍取决于跨细胞类型验证、覆盖度提升和成本下降。
Yang 的信号评级:High
理由:我会把这篇预印本的科研信号评为 High。它的强点不是某一个局部 biological finding,而是建立了一个可扩展的 deletion-scanning platform,并用相当大的规模证明它能同时读出 coding essentiality、noncoding purifying selection、essential region boundary 和 single-cell transcriptional phenotype。
这个 High 是“方法学和功能基因组学信号 High”,不是“临床就绪 High”。最值得关注的是 Shred-seq 可能把 ORF-centric functional genomics 推向 interval-centric functional genomics,让人类基因组的大片段结构、noncoding sequence 和 SV effect 能被系统性实验扰动。
但我会把应用成熟度评为 Medium-Low。原因是它仍是 preprint,主要验证在 HAP1 系统中完成,coverage 未饱和,scShred-seq 成本和统计功效仍有限。下一步最重要的不是再证明 Shred-seq 很酷,而是看它能否在更多 cell types、disease loci、stem-cell differentiation systems 和 targeted SV fine-mapping 中稳定产生可复现的 biological insight。
Jonas Koeppel and the team of Jay Shendure and Sudarshan Pinglay at the University of Washington, Seattle Hub for Synthetic Biology and Brotman Baty Institute recently posted a bioRxiv preprint introducing Shred-seq, a platform that couples CRISPR-Cas3 large deletions with phage polymerase-based breakpoint genotyping to systematically scan coding and noncoding intervals for cellular essentiality in the human genome.
What is the research question?
This paper asks a foundational functional genomics question: which contiguous DNA intervals in the human genome are required for cell survival and proliferation?
Until now, that question has mostly been answered at the ORF level. CRISPR knockout screens can systematically identify essential protein-coding genes, but the essentiality of the noncoding genome, multi-kilobase regulatory landscapes, large structural variants and broader genome architecture remains much harder to measure at scale. Comparative genomics, natural human structural variants and chromatin annotations provide indirect clues, but they do not directly answer what happens when a specific continuous stretch of DNA is deleted.
The central question of this preprint is therefore not whether one specific gene is essential. It is whether genome-scale deletion scanning can make coding, noncoding and large genomic intervals directly perturbable, directly genotypable and directly connectable to fitness or single-cell phenotypes.
What is truly new?
The real novelty of Shred-seq is that it connects two capabilities that normally live apart: Cas3 generates large, unidirectional and variable-length deletions; phage polymerase promoters turn unpredictable breakpoints into sequenceable genotype signals.
In practice, Shred-seq inserts beacons into the genome. Each beacon carries a unique barcode, a synthetic CRISPR target and T7/SP6 phage promoters. Type I-C CRISPR-Cas3 then initiates from the beacon and creates nested deletions that range from hundreds of base pairs to hundreds of kilobases. Because the deletion endpoint is not known in advance, conventional PCR primer design or clonal whole-genome sequencing does not scale well. Shred-seq solves that problem by using phage polymerase transcription from the beacon promoter to read out both the barcode and the newly formed genomic junction.
This turns deletion scanning from one locus at a time into a pooled screen across thousands of launchpads. Importantly, the same architecture can support bulk fitness readouts through in vitro transcription and single-cell phenotyping through in situ transcription plus scRNA-seq, creating scShred-seq.
Where is the data strongest?
The data are strongest because the authors do not stop at a conceptual method. They push the platform from proof of concept to gigabase-scale deletion scanning.
In the initial HAP1 proof of concept, the authors used PiggyBac to integrate Shred-seq beacons and recovered 10,744 barcodes, of which 6,549 could be mapped to unique genomic positions. After Cas3 editing, 34-36% of cells became GFP-negative, compared with 3.4% in control cells. T7-IVT breakpoint mapping identified 3,603 unique deletions in a single experiment, totaling 255 Mb and covering 151 Mb of the human reference genome. Deletion lengths showed a right-skewed long-tail distribution, with a median of about 33.1 kb, a maximum near 495 kb and roughly 24% longer than 100 kb.
The authors then optimized the beacon architecture. T7 promoter padding improved readout by 1.7-fold, while the v3 beacon with T7/SP6 non-convergent promoters improved readout by 18.3-fold relative to v1. They also compared PiggyBac, lentivirus and Sleeping Beauty integration preferences, showing that different delivery modalities bias launchpads toward different chromatin contexts and should be chosen based on the experimental question.
The main scale comes from four large Shred-seq screens. Combined, they recovered 36,257 deletions from 9,604 Cas3 launchpads, covering 461 Mb of unique sequence, or 13.9% of the human reference genome, with 2.55 Gb of cumulative deleted sequence. In terms of depth, 3.8% of the reference genome had at least five independent deletions and 1.8% had at least ten. A random-distribution model would produce roughly 200-fold less high-depth coverage.
The method also has mechanistic closure. Cas3-induced deletion lengths fit a log-normal distribution, with a fitted pre-selection median of 25.8 kb, close to the observed median of 27.5 kb. The authors mapped 7,467 deletion start sites and found the strongest initiation peak 22-34 nucleotides downstream of the protospacer, accounting for about 25.1% of deletions. At breakpoint junctions, 66.5% showed 1-10 bp microhomology, suggesting that MMEJ is the dominant repair pathway for Cas3 lesions; 19.2% showed insertions and 14.1% showed perfect junctions.
The key biological readout comes from selection over time. Across experiments with pre- and post-selection time points, 34,515 deletions covered 2.42 Gb of cumulative sequence. In haploid HAP1 cells, post-selection deletions were depleted for essential protein-coding genes; this shift disappeared in diploid HAP1 cells, consistent with buffering by a second allele. More importantly, even among 12,049 noncoding deletions that did not overlap protein-coding genes, post-selection deletions were depleted for active chromatin, A compartments, early replication, high GC content and some functional or constrained sequence features. This argues that selection is not driven only by ORFs.
Finally, scShred-seq shows that the platform can move beyond bulk viability. From 20,000 input cells, the authors generated matched transcriptome and T7 IST libraries, retained 13,944 single-cell profiles after QC, and focused on 9,951 cells with high T7 IST recovery. They assigned high-confidence deletion annotations to 4,680 cells. In a gene-dense region on chromosome X, RBM3-overlapping deletions significantly reduced RBM3 expression, and RBM3 became the strongest genome-wide differential expression hit, showing that deletion genotypes can be linked directly to transcriptional phenotypes.
What is the biggest weakness?
The biggest weakness is that this is still a bioRxiv preprint and has not yet been peer reviewed. The scale and method design are strong, but key claims still need review, independent replication and broader validation.
A second limitation is that the experiments are centered on HAP1, especially haploid or near-haploid cancer-derived cells. Cellular essentiality depends strongly on cell type, ploidy, growth condition and developmental state. A region that appears dispensable or constrained in HAP1 cannot be directly generalized to primary cells, stem cells, differentiated lineages or organismal viability.
A third limitation is that coverage is not yet truly genome-wide or saturating. Covering 461 Mb of unique sequence is substantial, but it is still about 14% of the human reference genome. The estimated lower and upper bounds for the dispensable fraction of the haploid genome, roughly 50% to 96%, are intentionally broad. At this stage, the main value is proof that the platform can work, not a final answer to the minimal human genome question.
A fourth limitation is engineering maturity. Flow sorting constrains scalability; random integration favors open or active chromatin, leaving heterochromatin and polycomb-repressed DNA under-covered; Cas3 deletion length distributions are not yet precisely programmable; and scShred-seq remains limited by cost, throughput and statistical power for subtle effects, rare phenotypes or combinatorial interactions.
Is there translational or clinical relevance?
This is not a clinical paper and should not be read as a direct therapeutic tool. Its translational relevance is platform-level.
First, it creates a possible route for functional fine-mapping of disease-associated structural variants. Many recurrent human disease SVs span multiple genes, enhancers and regulatory elements. A targeted version of Shred-seq could generate dense nested deletion series across those loci and help identify the causal subsequences driving phenotype.
Second, it could provide training data for noncoding genome maps and genome AI. Most current perturbation datasets are dominated by ORF-level Perturb-seq. Perturbation-phenotype pairs for noncoding intervals, large SVs and genome architecture remain scarce. Shred-seq can generate deletions of different lengths across different genomic contexts and connect them to fitness or transcriptome readouts, a data type that could matter for variant effect prediction, generative genome models and virtual cell models.
Third, it gives synthetic biology and minimal human genome design an experimental path. The authors discuss both top-down progressive deletion and bottom-up genome synthesis. That direction remains distant, but Shred-seq supplies a way to directly test which genomic intervals can be removed, rather than relying only on conservation or annotation.
In short, the translational meaning is closer to functional genomics infrastructure than clinical readiness. It may influence disease SV interpretation, synthetic genome design, cell engineering and AI training data, but real application maturity will depend on validation across cell types, higher coverage and lower-cost readouts.
Yang’s signal rating: High
Reason: I would rate the scientific signal of this preprint as High. The strength is not one local biological finding. It is the construction of a scalable deletion-scanning platform, with enough data to show that it can read out coding essentiality, noncoding purifying selection, essential-region boundaries and single-cell transcriptional phenotypes.
This High means method and functional-genomics signal High, not clinical readiness High. The most important implication is that Shred-seq may push functional genomics from an ORF-centric view toward an interval-centric view, making large genome structure, noncoding sequence and structural-variant effects experimentally perturbable at scale.
I would rate application maturity as Medium-Low. The work is still a preprint, the main validation is in HAP1, coverage is not saturating, and scShred-seq remains limited by cost and statistical power. The next decisive test is not whether Shred-seq is conceptually exciting, but whether it can produce reproducible biological insight across more cell types, disease loci, stem-cell differentiation systems and targeted SV fine-mapping experiments.