SEED | PRIME-In:让 CAR-T 大片段整合绕开 DSB SEED | PRIME-In inserts large DNA without DSBs AI-assisted · reviewed
中国科学院动物研究所、北京干细胞与再生医学研究院等机构的 Sen Fang、Na Tang、Yiyun Li、Shuyu Guo、Yangcan Chen 与 Haoyi Wang、Chenxin Wang、Wei Li 团队近期报道,PRIME-In 将 prime editor 生成的 donor primed microhomology flap 与 genomic nicking 结合,在不依赖 double-stranded DNA break repair 的情况下把 2-9.6 kb DNA 载荷整合进多种人细胞,并在原代人 T 细胞中实现高效 CD19 CAR knock-in 和高 genome-wide integration precision,为非病毒 CAR-T 制造提供了一条新的平台路径。
研究问题是什么?
这篇文章问的是一个 CAR-T 制造里的核心工艺问题:能否在原代人 T 细胞中非病毒、定点、高效地插入大 DNA 载荷,同时避免 DSB 带来的基因组安全风险和细胞扩增损失?
现有 CAR-T 产品主要依赖 lentiviral vector,把 transgene 随机整合进基因组。这个路线成熟、效率高,但随机整合带来的 insertional mutagenesis 风险、批间差异、成本和表达不一致,一直是产业化和安全性上的负担。
非病毒定点整合看起来更理想,但常见 HDR/HMEJ/CRISPR-Cas9 knock-in 需要在基因组靶点制造 DSB。DSB 可以提高插入效率,却也会带来 indel、reverse/multimeric insertion、chromosomal translocation、off-target integration、p53 activation 和 T cell death。对 CAR-T 来说,这不只是编辑“纯度”问题,也是最终活细胞产量和临床制造可行性问题。
因此,真正的问题不是简单提高 knock-in percentage,而是能否同时满足四件事:大载荷、定点整合、少基因组异常、在原代 T 细胞里还能保住足够扩增和功能。
真正的新意是什么?
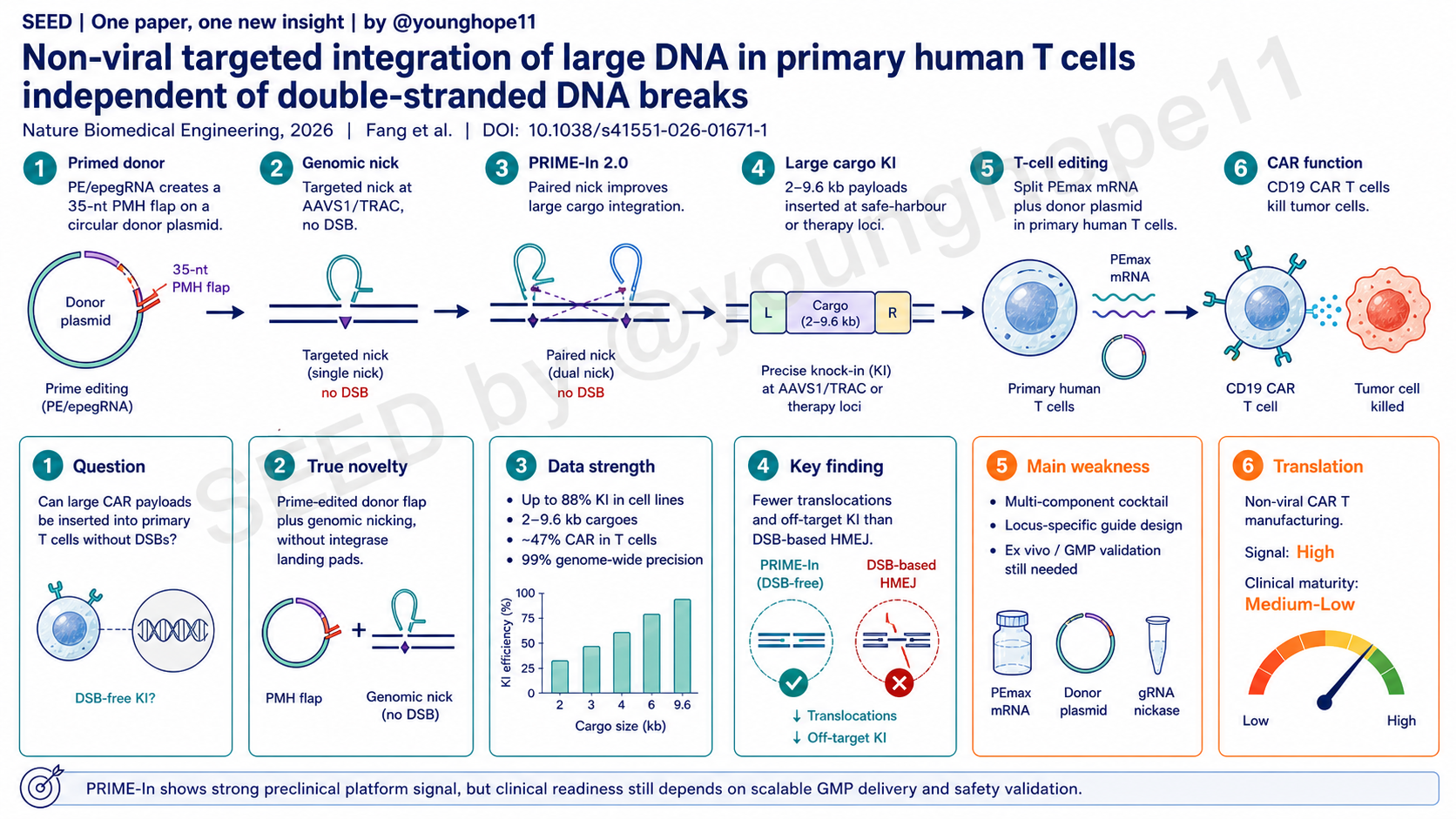
真正的新意是 PRIME-In 把 prime editing 的“精准 nick + 逆转录延伸”能力从直接改 genome,转移到 donor plasmid 处理上:prime editor 和 donor-targeting pegRNA/epegRNA 先在 donor 上生成 3’ primed microhomology flap,再由 gene-specific sgRNA 在基因组靶点制造 nick,让 donor flap 与 genomic nick 附近同源序列配对,随后通过 endogenous polymerase 延伸和 homology-based repair 完成大片段整合。
这个设计有两个关键点。
第一,donor 是多拷贝输入的 plasmid,作者把较复杂的 primed flap 生成放在 donor 上,而不是把所有压力放在单拷贝 genome target 上。这样可以增加 donor-genome annealing 的尝试机会,也绕开了传统 dsDNA donor 需要先寻找 DSB termini 的逻辑。
第二,PRIME-In 2.0 在基因组互补链下游增加一个 egRNA-directed nick。它不像 PE3 那样只在很近距离用第二个 nick 偏置修复,而是在几百 bp 间距下仍能提高大载荷整合效率。作者认为 long right homology arm 让这个远距离 paired nick 仍能帮助 heteroduplex resolution 和 downstream repair。
与 PASTE / PASSIGE / eePASSIGE 等 integrase-coupled prime editing 方法相比,PRIME-In 不需要先放入 landing pad,也不依赖 Bxb1-like recombination。与 HMEJ/HDR 相比,它避免了 nuclease-induced DSB。概念上,它更像是把“prime-edited donor flap + genomic nick”变成一种 DSB-free 的大 DNA integration interface。
数据强在哪里?
数据强在三个层级:机制可行性、编辑结果纯度、以及原代 T 细胞制造闭环。
第一层是 HEK293T 等细胞中的大载荷整合效率。作者用 2-kb EF1α-EGFP reporter 靶向 HEK4、HEK3、LSP1 和 VEGFA。PRIME-In 1.0 在四个位点分别达到 57.17%、19.43%、13.20% 和 22.13% EGFP+ stable integration,而 nicked HR donor、ldsDNA、lssDNA 或 covalent circular HR donor 整体低得多。优化 donor-targeting RT template 后,约 35 nt RT template 最优;再把 PE2/pegRNA 升级为 PEmax/epegRNA,并引入 PRIME-In 2.0 paired nick,最高在 HEK4 达到 88% knock-in,VEGFA 从 22.10% 提高到 83.57%。
第二层是载荷和靶点范围。PRIME-In 2.0 在 AAVS1、CCR5 等 safe-harbour loci 以及多个 therapy-related loci 上测试,能整合 2-9.6 kb 载荷。作者还展示了 CD19 CAR、IL2 CDS、GBA1 CDS 等治疗相关片段在 TRAC、CD3ζ、RGS16、GBA1 等位点的插入,说明它不是只适配一个 reporter 或一个 locus 的方法。
第三层是编辑纯度。PCR-enriched HTS 比较了 AAVS1 位点的 HMEJ、eePASSIGE、PRIME-In 1.0 和 PRIME-In 2.0。PRIME-In 1.0 和 2.0 的 intended KI 分别为 47.07% 和 59.43%,高于 HMEJ 的 23.70% 和 eePASSIGE 的 12.51%;editing errors 分别为 9.32% 和 14.47%,明显低于 HMEJ 的 56.98%。HMEJ 在 on-target site 产生 2.34% translocation,而 DSB-independent 方法只有 0.23-0.43%。在两个 predicted off-target cleavage sites,HMEJ 出现 off-target KI、indel 和 translocation,PRIME-In 1.0/2.0 则基本是 background-level events。Genome-wide insertion profiling 也显示 PRIME-In 的 off-target KI proportion 约 3.9%,明显低于 HMEJ 和 eePASSIGE。
最重要的是,作者把方法推到了原代人 T 细胞。通过 CMV-driven split PEmax、LinearDesign 优化 mRNA、低剂量 split PEmax mRNA 和 donor/gRNA 组合,PRIME-In 在 AAVS1 reporter setting 中实现高工程化 T cell yield。用 EF1α-EGFP benchmark 时,PRIME-In 的 EGFP+ T cell efficiency 为 31.27%,高于 HDR、HMEJ 和 ssCTS;DSB_PRIME-In 虽有相近 integration frequency,但 T cell recovery 更差,最终 engineered T cells 产量低于 PRIME-In。
T 细胞 DNA electroporation 的毒性也是重点。作者用 HCMV UL36/UL37x1 mRNA 和 A151 DNA sensor inhibitor 组合,显著提高 DNA challenge 后 T cell viability:UL36/UL37x1 从 33.67% 提升到 62.00%,再加 A151 后达到 81.00%。在 5.6-kb CD19 CAR-2A-EGFP cassette 中,没有 innate immune modifier 时 PRIME-In 约 20% integration,并在 7 天内得到约 3-fold engineered CAR T cells;UL36/UL37x1/A151 cocktail 又把 CAR T cell yield 额外提高约 3-fold。
最后,功能数据形成了相对完整的 CAR-T 闭环。AAVS1 EF1α-CD19 CAR-EGFP 的 PRIME-In integration rate 与 lentiviral transduction 接近,约 30.30% 对 30.53%。去掉 EGFP 后,AAVS1 EF1α-CD19 CAR 的 CAR expression 约 47%,7 天后 CAR T cell number 达到 11-fold over input,超过 lentiviral comparator。两名 donor 的 HTS 显示 PRIME-In-engineered T cells 有 high on-target efficiency、minimal errors 和 99% genome-wide integration precision。体外 Nalm6 killing、多轮 antigen exposure、cytokine staining,以及 Raji xenograft in vivo clearance 都显示 AAVS1-targeted PRIME-In CAR T cells 与 lentiviral 或 HMEJ 生成的 CAR T cells 具有可比功能。
最大弱点是什么?
最大弱点是 PRIME-In 的系统复杂度很高。一个完整 PRIME-In 2.0 cocktail 需要 PE 或 split PEmax、transgene-containing donor、donor-targeting epegRNA、genomic-targeting sgRNA,通常还需要 egRNA;在原代 T 细胞里,为了提高 viability 和 yield,还叠加了 split PEmax mRNA、UL36/UL37x1 mRNA 和 A151。这个组合在论文实验里有效,但走向 GMP manufacturing 时会带来原料、剂量窗口、释放检测、稳定性和法规路径的复杂性。
第二个弱点是 locus 和 guide design 依赖明显。作者筛了 48 个 genomic loci 并总结了 GC content、chromatin accessibility 等因素,但也承认不是所有位点都能达到同样高效率,新靶点仍需要经验性优化。对 CAR-T 产品开发来说,每换一个 locus、payload 或细胞来源,都可能需要重新建立设计和 QC。
第三个弱点是 safety 证据还主要来自短期、富集样本和 targeted HTS/NGS readout。文章显示 PRIME-In 比 DSB-based methods 更少 translocation 和 off-target KI,这是很强的相对安全信号;但临床级产品仍需要更长时间、更大样本、多 donor、多 batch 的验证,也需要 long-read sequencing、karyotype、WGS 或更系统的 integration-site safety assay。尤其 PRIME-In 2.0 有 200-400 bp moderate deletions 的 nick-out by-product,虽然不同于 DSB translocation,但仍需要在不同 loci 和 primary cell contexts 中持续评估。
第四个弱点是功能验证仍是 preclinical。CD19 CAR T cells 的 in vitro killing 和 Raji xenograft 数据足以说明 engineered cells 有功能,但 NXG xenograft 不能回答真实临床中的 persistence、exhaustion、immunogenicity、tumour heterogeneity、patient-to-patient variability 和 manufacturing failure rate。PRIME-In 能否在临床级 CAR-T 工艺里稳定超过 viral manufacturing,还没有被证明。
最后,UL36/UL37x1/A151 这一路线虽然很聪明,但也会成为转化中的审查重点。用病毒 anti-apoptosis/innate immune antagonist mRNA 暂时重编程 T cell stress response,有利于 yield,但必须证明不会改变 T cell long-term phenotype、genomic surveillance、tumorigenic risk 或 downstream immune behaviour。
是否有临床转化意义?
有,转化意义主要在非病毒 CAR-T manufacturing 平台。
如果 PRIME-In 能被放大到 GMP 级别,它可以同时解决几个长期痛点:减少 viral vector 依赖,降低随机整合风险,把 CAR 或其他 payload 放入 AAVS1、TRAC、CD3ζ、CD47 等预设位点,提高表达可控性,并有机会把 knock-in 与 knockout 组合到同一套 prime editing 平台中。对下一代 cell therapy 来说,这比单纯“更高 EGFP knock-in”重要得多。
它也提供了一个思路:DSB-free large-payload editing 不一定只能靠 transposase 或 integrase。通过 donor flap engineering 和 nick-based genome entry,prime editing 可以被重新组织成大 DNA insertion 工具。这个概念未来可能扩展到 TCR-T、Treg、NK cell、HSPC 或 iPSC-derived immune cell engineering。
但临床成熟度还不能高估。现在的数据证明的是 preclinical platform feasibility,而不是临床产品就绪。真正进入临床前,还需要回答 scalable manufacturing、closed-system delivery、batch consistency、donor variability、long-term integration safety、CAR expression stability、T cell phenotype preservation,以及监管机构能否接受 UL mRNA/A151 这类 yield-enhancing cocktail 等问题。
Yang 的信号评级:High
理由:我会把这篇论文的科研/平台信号评为 High。它提出了一个清晰的新机制界面,把 prime-edited donor flap 与 genomic nicking 结合,并用多层数据证明这不是一个只在 reporter system 中成立的小技巧:它覆盖 2-9.6 kb payload、多细胞系、多治疗相关 loci、HTS integration purity、原代 T 细胞 yield、CD19 CAR expression、体外杀伤和 in vivo xenograft function。
这个 High 是“科研信号 High”,不是“临床就绪 High”。PRIME-In 的相对安全性和高产量非常值得重视,但它仍是复杂的 ex vivo gene-editing manufacturing platform,离临床级产品还需要大量工艺、长期安全和法规验证。
临床成熟度我会评为 Medium-Low。它已经比许多 DSB-free large-payload insertion 概念更接近真实 T cell manufacturing,但还没有进入 patient-scale GMP 和人体安全/疗效验证阶段。下一步最关键的是证明这套多组件体系能在临床制造条件下稳定、可控、可质检,并且在多 donor、多 batch 中保持低 off-target integration 和良好 CAR-T 功能。
Sen Fang, Na Tang, Yiyun Li, Shuyu Guo, Yangcan Chen and the Haoyi Wang, Chenxin Wang and Wei Li team at the Institute of Zoology, Chinese Academy of Sciences, Beijing Institute for Stem Cell and Regenerative Medicine and collaborating institutions recently reported PRIME-In, a platform that couples a prime-editor-generated donor primed microhomology flap with genomic nicking to integrate 2-9.6 kb DNA payloads into human cells without relying on double-stranded DNA break repair, and that enables efficient CD19 CAR knock-in with high genome-wide integration precision in primary human T cells.
What is the research question?
This paper asks a core manufacturing question for CAR-T engineering: can large DNA payloads be inserted into primary human T cells in a non-viral, site-specific and efficient way while avoiding the genome-safety and cell-expansion costs of DSBs?
Current CAR-T products still largely rely on lentiviral vectors, which randomly integrate the transgene into the genome. This route is mature and efficient, but random integration brings concerns about insertional mutagenesis, batch-to-batch variability, cost and inconsistent transgene expression.
Non-viral targeted integration is attractive, but common HDR, HMEJ and CRISPR-Cas9 knock-in approaches create DSBs at the genomic target. DSBs can improve insertion efficiency, but they also bring indels, reverse or multimeric insertions, chromosomal translocations, off-target integration, p53 activation and T-cell death. For CAR-T manufacturing, this is not only an editing-purity issue. It directly affects viable engineered-cell yield and clinical manufacturing feasibility.
The real question is therefore not simply how to raise the knock-in percentage. It is whether one method can satisfy four requirements at once: large cargo, targeted integration, fewer genomic abnormalities and enough primary T-cell recovery and function.
What is truly new?
The real novelty is that PRIME-In redirects the precision nicking and reverse-transcription logic of prime editing away from directly rewriting the genome and toward processing the donor plasmid. A prime editor and donor-targeting pegRNA or epegRNA first generate a 3’ primed microhomology flap on the donor. A gene-specific sgRNA then creates a genomic nick, allowing the donor flap to anneal near the nicked genomic target, followed by endogenous polymerase extension and homology-based repair to complete large-payload integration.
Two design choices matter.
First, the donor is a multicopy input plasmid. PRIME-In places the more complex primed-flap generation step on the donor rather than forcing every step to occur at a single-copy genomic target. This increases donor-genome annealing opportunities and avoids the classic logic of dsDNA donors needing to find DSB termini.
Second, PRIME-In 2.0 adds an egRNA-directed nick on the downstream complementary genomic strand. Unlike PE3, where the second nick is usually close to the edit, PRIME-In 2.0 can gain efficiency even with nick distances of hundreds of base pairs. The authors argue that the long right homology arm enables this distant paired nick to help heteroduplex resolution and downstream repair.
Compared with PASTE, PASSIGE or eePASSIGE-like integrase-coupled prime-editing systems, PRIME-In does not require landing-pad insertion and does not rely on Bxb1-like recombination. Compared with HMEJ or HDR, it avoids nuclease-induced DSBs. Conceptually, it turns a prime-edited donor flap plus a genomic nick into a DSB-free large-DNA integration interface.
Where is the data strongest?
The data are strongest at three levels: mechanistic feasibility, editing-outcome purity and a primary T-cell manufacturing loop.
The first level is large-payload integration in HEK293T and other human cells. Using a 2-kb EF1α-EGFP reporter targeted to HEK4, HEK3, LSP1 and VEGFA, PRIME-In 1.0 achieved stable EGFP+ integration frequencies of 57.17%, 19.43%, 13.20% and 22.13%, respectively. Nicked HR donors, ldsDNA, lssDNA and covalent circular HR donors performed substantially worse. After optimizing donor-targeting RT-template length, an approximately 35-nt RT template was best. Upgrading PE2/pegRNA to PEmax/epegRNA and adding PRIME-In 2.0 paired nicking drove peak knock-in to 88% at HEK4 and raised VEGFA integration from 22.10% to 83.57%.
The second level is cargo and target breadth. PRIME-In 2.0 was tested at safe-harbour loci such as AAVS1 and CCR5 and across multiple therapy-related loci, with payloads of 2-9.6 kb. The paper also shows insertion of therapeutic sequences such as CD19 CAR, IL2 CDS and GBA1 CDS at TRAC, CD3ζ, RGS16, GBA1 and other sites. This makes the method more than a single-reporter, single-locus result.
The third level is editing purity. PCR-enriched HTS compared HMEJ, eePASSIGE, PRIME-In 1.0 and PRIME-In 2.0 at AAVS1. PRIME-In 1.0 and 2.0 achieved intended KI frequencies of 47.07% and 59.43%, higher than HMEJ at 23.70% and eePASSIGE at 12.51%. Editing errors were 9.32% and 14.47% for PRIME-In 1.0 and 2.0, far below HMEJ at 56.98%. HMEJ produced 2.34% translocations at the on-target site, whereas the DSB-independent methods remained at 0.23-0.43%. At two predicted off-target cleavage sites, HMEJ generated off-target KI, indels and translocations, while PRIME-In 1.0 and 2.0 showed only background-level events. Genome-wide insertion profiling also showed PRIME-In off-target KI proportions of about 3.9%, substantially lower than HMEJ and eePASSIGE.
Most importantly, the authors pushed the method into primary human T cells. With CMV-driven split PEmax, LinearDesign-optimized mRNA, low-dose split PEmax mRNA and a combined donor/gRNA setup, PRIME-In generated high engineered T-cell yield in an AAVS1 reporter setting. In an EF1α-EGFP benchmark, PRIME-In produced 31.27% EGFP+ T cells, outperforming HDR, HMEJ and ssCTS. DSB_PRIME-In reached a similar integration frequency, but T-cell recovery was worse, leaving final engineered-cell yield below PRIME-In.
DNA electroporation toxicity in T cells is another key piece of the paper. The authors used HCMV UL36/UL37x1 mRNAs and the A151 DNA-sensor inhibitor to improve T-cell viability under DNA challenge. UL36/UL37x1 increased viability from 33.67% to 62.00%, and adding A151 raised it to 81.00%. For a 5.6-kb CD19 CAR-2A-EGFP cassette, PRIME-In without innate immune modifiers achieved about 20% integration and roughly 3-fold engineered CAR T cells over input after 7 days. The UL36/UL37x1/A151 cocktail then increased CAR T-cell yield by an additional approximately 3-fold.
Finally, the CAR-T functional dataset closes a meaningful loop. PRIME-In-mediated AAVS1 EF1α-CD19 CAR-EGFP integration was comparable to lentiviral transduction, at 30.30% versus 30.53%. Removing EGFP, AAVS1 EF1α-CD19 CAR expression reached about 47%, and CAR T-cell numbers reached 11-fold over input after 7 days, surpassing the lentiviral comparator. HTS from two donors showed high on-target efficiency, minimal errors and 99% genome-wide integration precision in PRIME-In-engineered T cells. In vitro Nalm6 killing, repeated antigen stimulation, cytokine staining and Raji xenograft clearance showed that AAVS1-targeted PRIME-In CAR T cells were functionally comparable to lentiviral or HMEJ-generated CAR T cells.
What is the biggest weakness?
The biggest weakness is system complexity. A full PRIME-In 2.0 cocktail requires PE or split PEmax, a transgene-containing donor, a donor-targeting epegRNA, a genomic-targeting sgRNA and often an egRNA. In primary T cells, the authors further add split PEmax mRNA, UL36/UL37x1 mRNAs and A151 to improve viability and yield. This cocktail works in the paper, but GMP manufacturing would have to handle reagent sourcing, dose windows, release assays, stability and regulatory complexity.
A second weakness is locus and guide-design dependence. The authors screened 48 genomic loci and identified factors such as GC content and chromatin accessibility, but they also acknowledge that not every target site reaches the same efficiency and that new loci may still require empirical optimization. For CAR-T product development, each new locus, payload or cell source may require a new design and QC package.
A third weakness is that the safety evidence is still mainly short-term and based on enriched samples plus targeted HTS and NGS readouts. The paper provides a strong relative safety signal against DSB-based methods, with fewer translocations and off-target KI events. But clinical-grade products will need longer-duration, larger-sample, multi-donor and multi-batch validation, ideally including long-read sequencing, karyotyping, WGS or more systematic integration-site safety assays. PRIME-In 2.0 also produces 200-400 bp moderate deletions through a nick-out by-product. These are different from DSB-driven translocations, but still need to be followed across loci and primary-cell contexts.
A fourth weakness is that functional validation remains preclinical. The in vitro killing and Raji xenograft data are enough to show that the engineered cells can function, but an NXG xenograft does not answer clinical questions about persistence, exhaustion, immunogenicity, tumor heterogeneity, patient-to-patient variability or manufacturing failure rate. PRIME-In has not yet shown that it can reliably outperform viral manufacturing in a clinical-grade CAR-T process.
Finally, the UL36/UL37x1/A151 strategy is clever but will draw translational scrutiny. Transiently using viral anti-apoptosis and innate-immune antagonist mRNAs to reprogram the T-cell stress response may improve yield, but developers will need to prove that it does not alter long-term T-cell phenotype, genomic surveillance, tumorigenic risk or downstream immune behavior.
Is there translational or clinical relevance?
Yes. The translational relevance is primarily in non-viral CAR-T manufacturing.
If PRIME-In can be scaled into a GMP-grade process, it could address several long-standing bottlenecks at once: reduce dependence on viral vectors, lower random-integration risk, place CARs or other payloads into predefined sites such as AAVS1, TRAC, CD3ζ or CD47, improve transgene-expression control and potentially combine knock-in and knockout operations within a prime-editing-based platform. For next-generation cell therapy, that matters more than simply achieving a higher EGFP knock-in percentage.
It also broadens how the field can think about DSB-free large-payload editing. Large DNA insertion may not have to rely only on transposases or integrases. By engineering the donor flap and using a nick-based genomic entry point, prime editing can be reorganized into a large-DNA insertion tool. The same logic could potentially extend to TCR-T, Treg, NK-cell, HSPC or iPSC-derived immune-cell engineering.
But clinical maturity should not be overestimated. The current data demonstrate preclinical platform feasibility, not product readiness. Before clinical translation, the key questions will be scalable manufacturing, closed-system delivery, batch consistency, donor variability, long-term integration safety, CAR-expression stability, T-cell phenotype preservation and whether regulators will accept yield-enhancing components such as UL mRNAs and A151.
Yang’s signal rating: High
Rationale: I would rate the scientific and platform signal as High. The paper proposes a clear new mechanistic interface by coupling a prime-edited donor flap with genomic nicking, then supports it across multiple layers: 2-9.6 kb payloads, several human cell lines, multiple therapy-related loci, HTS integration purity, primary T-cell yield, CD19 CAR expression, in vitro killing and in vivo xenograft function.
This High means high scientific signal, not high clinical readiness. PRIME-In’s relative safety and high engineered-cell yield are important, but it is still a complex ex vivo gene-editing manufacturing platform that needs substantial process, long-term safety and regulatory validation.
I would rate clinical maturity as Medium-Low. PRIME-In is closer to real T-cell manufacturing than many DSB-free large-payload insertion concepts, but it has not yet entered patient-scale GMP manufacturing or human safety and efficacy testing. The decisive next step is showing that this multicomponent system remains stable, controllable and quality-checkable under clinical manufacturing conditions, while preserving low off-target integration and strong CAR-T function across multiple donors and batches.