SEED | 工程化 HSPC,把 IFNα 送进 GBM 冷肿瘤 SEED | Engineered HSPCs carry IFNα into cold GBM AI-assisted · reviewed
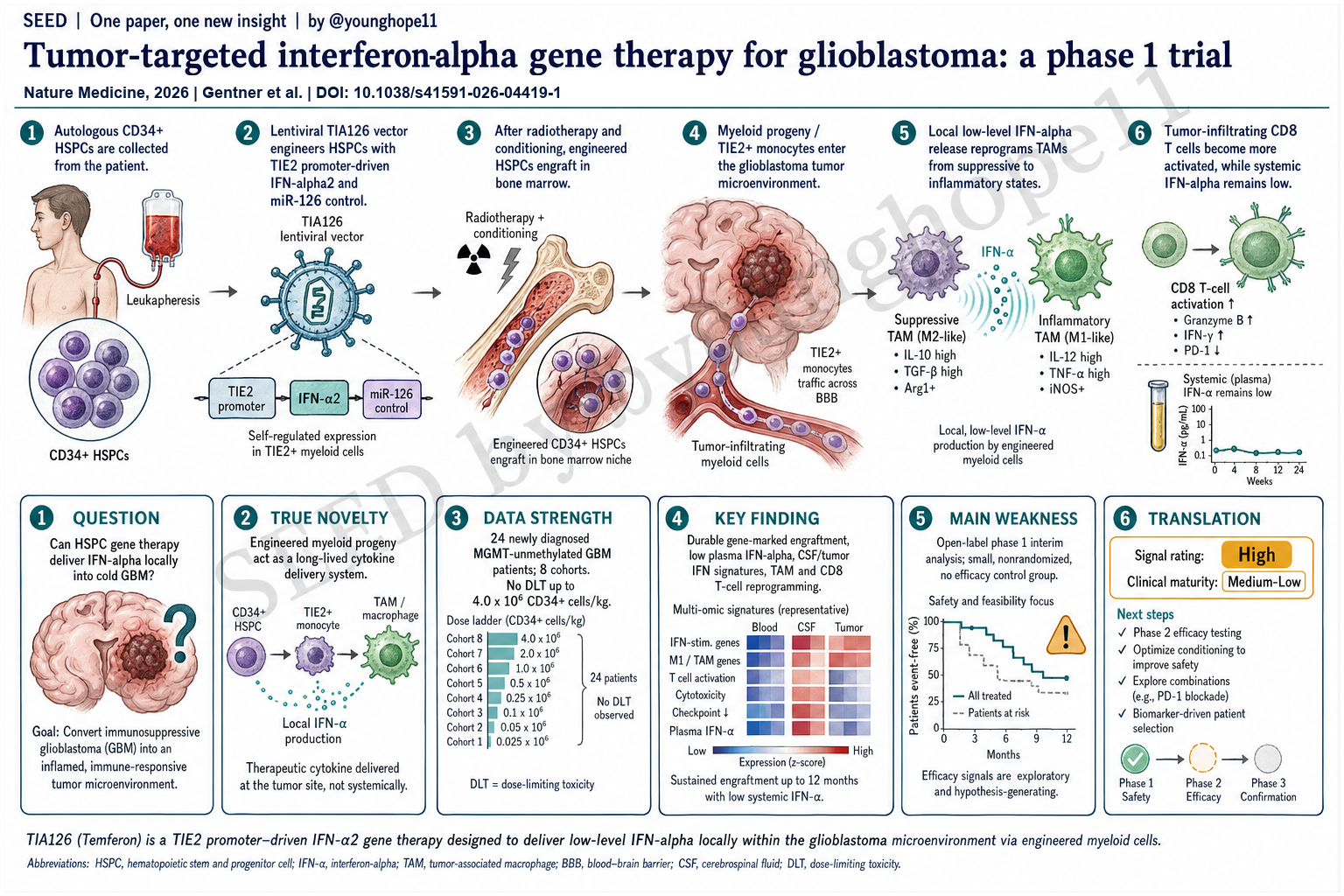
San Raffaele Telethon Institute for Gene Therapy、San Raffaele Hospital 和合作临床中心的 Bernhard Gentner 与 Fabio Ciceri、Luigi Naldini 团队近期报道,工程化自体 CD34+ HSPC 可通过髓系后代进入 glioblastoma tumor microenvironment,局部释放 IFNα2,并在新诊断、MGMT promoter 未甲基化 GBM 的 phase 1/2a 剂量递增试验中显示可行性、安全性和持续生物学活性,为用 HSPC gene therapy 重塑实体瘤髓系微环境提供了早期临床证据。
研究问题是什么?
这篇文章问的是一个 GBM 免疫治疗和细胞治疗交叉处的问题:能不能用工程化造血干/祖细胞,持续生成带有抗肿瘤功能的髓系后代,把 IFNα 局部送进 glioblastoma 这种免疫冷、髓系抑制强的肿瘤微环境?
GBM 对 checkpoint blockade 等免疫治疗普遍反应差,一个核心原因是 TME 中 tumor-associated macrophages 和其他髓系细胞占主导,形成免疫抑制、促血管生成和促侵袭状态。直接系统性给 IFN 或其他炎症性细胞因子,理论上能激活抗肿瘤免疫,但毒性和药代动力学会限制持续暴露。
这篇论文的核心问题因此不是“IFNα 是否能刺激免疫”,而是能否建立一种更精准的递送逻辑:让 HSPC 长期植入骨髓,持续产生会被肿瘤招募的工程化 myeloid progeny;这些细胞到达 GBM TME 后,在 TIE2/TEK 相关髓系状态下局部表达 IFNα2,从而避免高系统暴露,同时把髓系和 T 细胞生态推向更炎症、更抗肿瘤的方向。
真正的新意是什么?
真正的新意是把 HSPC gene therapy 从遗传病领域的“长期补酶/补基因”逻辑,转成实体瘤免疫治疗中的“长期、内源性、肿瘤靶向 cytokine delivery system”。
Temferon 的设计很巧。患者自体 CD34+ HSPC 经 lentiviral TIA126 vector 转导,IFNα2 由 TIE2 enhancer/promoter 驱动,使表达偏向 TIE2+ tumor-infiltrating myeloid cells;同时 vector 中加入 miR-126 target sequences,在 HSPC 中压低转基因表达,减少骨髓内或系统性 IFNα 暴露。也就是说,它不是把 IFNα 直接输进全身,而是把“产生 IFNα 的能力”装进能长期植入并分化为髓系细胞的 HSPC。
这与成熟单核/巨噬细胞治疗不同。成熟 MoMac 寿命、增殖和体内稳定性有限;HSPC 则能长期在骨髓中维持,持续补充能进入肿瘤的髓系后代。如果这个逻辑成立,它代表的是一种新的实体瘤细胞治疗平台:用 HSPC 作为长期制造和分发工程化髓系细胞的源头。
数据强在哪里?
数据最强的地方在于它完成了 first-in-human 安全性、植入、组织进入和机制读出的闭环,而不是只停留在可制造性。
临床设计上,TEM-GBM_001 是开放标签、多中心、phase 1/2a、剂量递增研究。Part A 报告了 24 名实际接受 Temferon 的新诊断、MGMT promoter 未甲基化 GBM 患者,分布于 8 个 cohort。Temferon 剂量从 0.5 × 10^6 到 4.0 × 10^6 CD34+ cells/kg;conditioning 比较了 BCNU/thiotepa、busulfan/thiotepa 和 busulfan monotherapy。主要终点是 infusion 后 90 天内安全性和耐受性。
安全性方面,到最高剂量 4.0 × 10^6 cells/kg 未观察到 dose-limiting toxicity。Temferon infusion 本身耐受良好,未见归因于输注本身的不良事件;大多数 treatment-emergent adverse events 与 autologous stem cell transplant 和 conditioning chemotherapy 预期毒性一致。值得注意的是,双烷化剂 conditioning 组有 3 名患者在 4 个月内死于感染并发症,说明这一路线的临床风险并不轻;但 busulfan monotherapy 组感染和总体不良事件较少,因此被选择为后续开发方向。
植入和基因标记证据也较强。所有接受 Temferon 的患者都在 peripheral blood 和 bone marrow 中出现 gene-marked cells。CD33+ BM progenitors 中的 VCN 在 day +30 达峰,并与输入转导细胞比例相符;转导髓系细胞长期存在,随后下降并在多数患者中稳定。integration site analysis 未见 insertional mutagenesis 或 clonal dominance 信号。
关键机制证据来自“低系统暴露 + 局部活性”。尽管个别患者 blood leukocytes gene marking 可达 40-50%,plasma IFNα 在前 3 个月始终维持低水平,低于 31 pg/ml,且与 VCN 无明显相关。相反,CSF 中 IFNα 在治疗后逐步可测:day +30 为 6/12,day +90 为 11/15,day +180 和 day +360 时所有可评估患者均可测。复发或再手术肿瘤样本中,除最低 engraftment 的 cohort 1 患者外,均检测到 vector copies,tumor-derived DNA VCN 为 1-6%;CD45+ tumor digests 与匹配 PB CD14+ monocytes 比较显示,肿瘤白细胞中有相当比例来自 Temferon graft。
最有说服力的是 TME 机制读出。作者对 Temferon 后肿瘤样本和 SoC recurrent GBM 对照做了 scRNA-seq,并整合公开数据。Temferon-exposed tumors 中,IFN 和 inflammatory pathways 在肿瘤生态系统多个 compartment 中富集;髓系 compartment 从 M2-like / MIF+ / APOC+ 状态向 inflammatory macrophage 状态偏移;同时 GZMK+ CD8 effector、stem-like/activated T cells 和可能 tumor-reactive CD8 TILs 增加。患者 TEM-G11 的同体内比较尤其有价值:稳定病灶 TL1 有更高 IRG、更多 TEM、更多炎症 TAM 和更多 predicted tumor-reactive CD8 T cells;进展病灶 TL2 则相反。
临床信号则应保守解读。中位随访 39 个月时,6 个月未见 objective responses,但多数可评估患者为 stable disease;有两名长期生存者后期出现 partial response。中位 OS 和 PFS 分别为诊断后 16.7 个月和 8.1 个月,患者多数在一年内维持较好 performance status 和 quality of life。对 MGMT 未甲基化 GBM 来说,这是值得关注的早期信号,但还不能证明疗效。
最大弱点是什么?
最大弱点是这是 phase 1 interim analysis,核心设计是安全性和剂量/conditioning 选择,不是疗效验证。研究样本小,只有 24 名 dosed patients;开放标签、非随机、没有前瞻性对照组;8 个 cohort 同时改变 Temferon dose、supporter cell dose 和 conditioning regimen,使疗效或机制差异很难归因到单一变量。
第二个弱点是临床获益仍不确定。文章没有在 +6 个月观察到 objective response,中位 OS 16.7 个月虽然与 contemporary MGMT-unmethylated GBM datasets 相比有一定吸引力,但缺少随机对照,不能排除 patient selection、治疗路径和后续治疗的影响。两例 late partial responses 很有意思,但不足以定义疗效。
第三个弱点是 safety burden 不低。Temferon 本身未出现 DLT 是重要结果,但整个治疗流程包括 HSPC mobilization、apheresis、radiotherapy、conditioning、autologous transplant 和长期随访。双烷化剂 conditioning 下的 transplant-related mortality 提醒我们,这不是一个轻量级免疫治疗。未来如果要进入更广泛实体瘤或更脆弱患者人群,conditioning 毒性会是关键限制。
第四个弱点是机制证据多为 exploratory / post hoc,且组织样本主要来自 recurrence 或 second surgery。scRNA-seq 和 TCR evidence 很漂亮,但它们是在有限样本中、治疗后可取得组织的患者里完成,存在取样偏倚;而且 progression 样本可能低估或扭曲真实 immune engagement。TEM-G11 的同体内病灶比较很有解释力,但毕竟是单个患者的深描。
最后,论文存在明确产业相关性。Genenta Science 是研究 sponsor,参与预设终点的设计、执行和分析;Bernhard Gentner 和 Luigi Naldini 是 Genenta cofounders/shareholders。利益冲突不否定数据,但在解读 efficacy signal 和下一步开发叙事时需要保持独立判断。
是否有临床转化意义?
有,而且转化意义主要在两个层面。
第一层是 GBM 治疗本身。Temferon 提供了一种不同于 CAR-T 或 checkpoint blockade 的思路:不是直接攻击一个肿瘤抗原,而是持续改写 myeloid-dominant TME,让肿瘤局部更适合抗肿瘤免疫发生。对于 antigen heterogeneity 强、T cell exclusion 明显、髓系抑制突出的 GBM,这个方向有合理性。它也可能成为 CAR-T、checkpoint inhibitor、anti-VEGF 或其他局部免疫激活策略的组合伙伴。
第二层更重要:HSPC 作为实体瘤免疫治疗平台。若工程化 HSPC 可以稳定植入、长期产生进入实体瘤的髓系后代,并在局部释放可调 payload,那么未来 payload 不一定局限于 IFNα,也可能是 bispecific T-cell engagers、macrophage-activating receptors、checkpoint-modulating molecules 或其他免疫调节因子。这篇论文真正打开的是“用造血系统持续制造肿瘤靶向髓系细胞”的平台想象。
但它离临床就绪还远。下一步关键不是再证明 Temferon 可以植入,而是要在 phase 2 中证明有清楚的 survival 或 durable disease-control benefit;同时要优化 conditioning,降低感染和 transplant burden;还要建立 biomarker,识别哪些患者能获得足够 TEM recruitment、TIE2 promoter activity 和 IFN responsiveness。
Yang 的信号评级:High
理由:我会把这篇论文的科研/平台信号评为 High。它不是一个单纯 phase 1 安全性报告,而是把 HSPC engineering、髓系肿瘤归巢、局部 IFNα 递送、长期植入、低系统暴露、CSF/tumor IFN activity、single-cell TME reprogramming 和 TCR clonotype evidence 串成了一条相对完整的早期临床证据链。
这个 High 是“科研和平台信号 High”,不是“临床就绪 High”。Temferon 对 GBM 是否真正改善生存,还需要随机或至少更有说服力的 phase 2 数据;conditioning 负担和感染风险也必须被显著优化。
临床成熟度我会评为 Medium-Low。它已经跨过 first-in-human feasibility 和安全性第一关,并有机制读出支持,但目前仍是小样本、非随机、interim phase 1 结果。真正决定价值的下一步,是看 busulfan-based regimen 下的推荐 phase 2 dose、患者选择 biomarker 和 survival endpoint 能否同时站住。
Bernhard Gentner and the Fabio Ciceri and Luigi Naldini team at the San Raffaele Telethon Institute for Gene Therapy, San Raffaele Hospital and collaborating clinical centers recently reported that engineered autologous CD34+ HSPCs can generate myeloid progeny that enter the glioblastoma tumor microenvironment, locally release IFNα2 and show feasibility, safety and sustained biological activity in a phase 1/2a dose-escalation trial in newly diagnosed MGMT promoter-unmethylated GBM, providing early clinical evidence for using HSPC gene therapy to reprogram the myeloid ecosystem of solid tumors.
What is the research question?
This paper asks a question at the intersection of GBM immunotherapy and cell therapy: can engineered hematopoietic stem and progenitor cells durably generate antitumor myeloid progeny that deliver IFNα locally into the immunologically cold, myeloid-suppressed glioblastoma tumor microenvironment?
GBM generally responds poorly to immune checkpoint blockade and other immunotherapies. A central reason is that tumor-associated macrophages and other myeloid populations dominate the TME and create an immunosuppressive, proangiogenic and pro-invasive state. Systemic IFN or other inflammatory cytokines could, in principle, activate antitumor immunity, but toxicity and pharmacokinetics limit sustained exposure.
The core question is therefore not simply whether IFNα can stimulate immunity. It is whether HSPCs can be used as a durable, more precise delivery system: engraft in bone marrow, continuously generate engineered myeloid progeny recruited into GBM, express IFNα2 locally under a TIE2/TEK-associated myeloid program, avoid high systemic exposure and push the local myeloid and T-cell ecology toward a more inflammatory antitumor state.
What is truly new?
The real novelty is the conversion of HSPC gene therapy from a long-term enzyme or gene-replacement logic, familiar from inherited disorders, into a durable, endogenous, tumor-targeted cytokine delivery system for solid-tumor immunotherapy.
Temferon is designed around this idea. Autologous CD34+ HSPCs are transduced with the lentiviral TIA126 vector. IFNα2 is driven by a TIE2 enhancer/promoter, biasing expression toward TIE2+ tumor-infiltrating myeloid cells, while miR-126 target sequences suppress transgene expression in HSPCs to reduce bone-marrow and systemic IFNα exposure. In other words, the treatment does not simply infuse IFNα into the body. It installs the capacity to produce IFNα into HSPCs that can engraft long term and differentiate into tumor-infiltrating myeloid cells.
This differs from mature monocyte or macrophage cell therapy. Mature MoMacs have limited lifespan, proliferation and in vivo stability. HSPCs can persist in the marrow and continuously replenish myeloid progeny capable of entering tumors. If this logic holds, it points to a new class of solid-tumor cell therapy: using HSPCs as the long-term source for engineered myeloid-cell production and distribution.
Where is the data strongest?
The strongest part of the dataset is that it closes an early first-in-human loop across safety, engraftment, tumor entry and mechanism, rather than stopping at manufacturability.
Clinically, TEM-GBM_001 is an open-label, multicenter, phase 1/2a dose-escalation study. Part A reports 24 dosed patients with newly diagnosed, MGMT promoter-unmethylated GBM across 8 cohorts. Temferon doses ranged from 0.5 x 10^6 to 4.0 x 10^6 CD34+ cells/kg, and conditioning regimens included BCNU/thiotepa, busulfan/thiotepa and busulfan monotherapy. The primary endpoint was safety and tolerability during the first 90 days after infusion.
On safety, no dose-limiting toxicity was observed up to 4.0 x 10^6 cells/kg. Temferon infusion itself was well tolerated, with no adverse events attributed to the infusion. Most treatment-emergent adverse events were consistent with expected toxicities of autologous stem cell transplant and conditioning chemotherapy. Importantly, three patients in the double-alkylator conditioning cohorts died from infectious complications within 4 months, showing that the overall treatment burden is substantial. Busulfan monotherapy had fewer infections and fewer overall adverse events and was selected for further development.
Engraftment and gene-marking data are also strong. All Temferon-treated patients engrafted with gene-marked cells in peripheral blood and bone marrow. VCN in CD33+ BM progenitors peaked at day +30 and matched the inferred input proportion of transduced cells. Transduced myeloid cells persisted long term, then declined and stabilized in most patients. Integration-site analysis did not show insertional mutagenesis or clonal dominance.
The key mechanistic evidence is the combination of low systemic exposure and local activity. Even with blood-leukocyte gene marking reaching 40-50% in one patient, plasma IFNα remained consistently low during the first 3 months, below 31 pg/ml, with no clear correlation to VCN. In contrast, CSF IFNα became progressively detectable after treatment: 6 of 12 evaluable patients at day +30, 11 of 15 at day +90 and all evaluable patients at days +180 and +360. In recurrent or second-surgery tumor samples, vector copies were detected in all evaluable tumors except one low-engraftment cohort 1 patient, with tumor-derived DNA VCN of 1-6%. Comparing CD45+ tumor digests with matched PB CD14+ monocytes suggested substantial replenishment of tumor leukocytes from the Temferon graft.
The most persuasive biology comes from the TME readouts. The authors performed scRNA-seq on post-Temferon tumors and standard-of-care recurrent GBM controls, then integrated public datasets. Temferon-exposed tumors showed enrichment of IFN and inflammatory pathways across multiple tumor-ecosystem compartments. The myeloid compartment shifted away from M2-like, MIF+ and APOC+ states toward inflammatory macrophage states. At the same time, GZMK+ CD8 effectors, stem-like/activated T cells and putative tumor-reactive CD8 TILs increased. The intrapatient comparison in TEM-G11 is especially informative: the stable lesion TL1 had higher IRG expression, more TEMs, more inflammatory TAMs and more predicted tumor-reactive CD8 T cells, whereas the progressing TL2 lesion showed the opposite pattern.
The clinical signal should still be interpreted cautiously. At a median follow-up of 39 months, no objective responses were observed at 6 months, but most evaluable patients had stable disease. Two long-term survivors later achieved partial responses. Median OS and PFS from diagnosis were 16.7 and 8.1 months, respectively, with most patients maintaining good performance status and quality of life during the first year. For MGMT-unmethylated GBM, this is worth attention, but it does not prove efficacy.
What is the biggest weakness?
The biggest weakness is that this is a phase 1 interim analysis. The study is built primarily for safety, dose selection and conditioning-regimen selection, not efficacy validation. The sample size is small, with 24 dosed patients; the trial is open-label and nonrandomized; there is no prospective control group; and the 8 cohorts changed Temferon dose, supporter-cell dose and conditioning regimen, making clinical or mechanistic differences hard to attribute to a single variable.
A second weakness is that clinical benefit remains uncertain. The study did not observe objective responses at 6 months. Median OS of 16.7 months is encouraging relative to contemporary MGMT-unmethylated GBM datasets, but without randomization it cannot separate treatment effect from patient selection, treatment path or subsequent therapies. The two late partial responses are interesting, but not enough to define efficacy.
A third weakness is the safety burden. The absence of Temferon-attributed DLT is important, but the full treatment path includes HSPC mobilization, apheresis, radiotherapy, conditioning, autologous transplant and long-term follow-up. The transplant-related mortality seen with double-alkylator conditioning is a reminder that this is not a lightweight immunotherapy. Conditioning toxicity will be a central limitation if this platform is moved into broader solid-tumor settings or more fragile patient populations.
Fourth, much of the mechanistic evidence is exploratory or post hoc, and tissue sampling mainly comes from recurrence or second surgery. The single-cell and TCR data are compelling, but they come from limited samples and from patients in whom post-treatment tissue was available, creating sampling bias. Progression samples may also underestimate or distort true immune engagement. The TEM-G11 intrapatient comparison is mechanistically rich, but it remains a deep case study in one patient.
Finally, the paper has clear industry involvement. Genenta Science sponsored the study and was involved in the design, conduct and analysis of prespecified endpoints. Bernhard Gentner and Luigi Naldini are Genenta cofounders and shareholders. These conflicts do not invalidate the data, but efficacy claims and development narratives should be interpreted independently.
Is there translational or clinical relevance?
Yes. The translational relevance operates on two levels.
The first is GBM therapy itself. Temferon offers a strategy different from CAR T cells or checkpoint blockade: instead of directly attacking a tumor antigen, it tries to durably rewrite a myeloid-dominated TME so that antitumor immunity can occur locally. For GBM, with strong antigen heterogeneity, T-cell exclusion and myeloid suppression, that is a rational direction. Temferon may also become a combination partner for CAR T cells, checkpoint inhibitors, anti-VEGF therapy or other local immune-activation strategies.
The second and more important level is HSPCs as a solid-tumor immunotherapy platform. If engineered HSPCs can stably engraft, continuously generate myeloid progeny that enter solid tumors and locally release tunable payloads, future payloads need not be limited to IFNα. They could include bispecific T-cell engagers, macrophage-activating receptors, checkpoint-modulating molecules or other immunoregulatory factors. The platform idea opened by this paper is the use of the hematopoietic system as a durable factory for tumor-targeted myeloid cells.
But it is still far from clinically ready. The next key step is not simply proving that Temferon engrafts. It is showing a clear survival or durable disease-control benefit in phase 2, while reducing conditioning toxicity and transplant burden, and building biomarkers that identify patients with sufficient TEM recruitment, TIE2 promoter activity and IFN responsiveness.
Yang’s signal rating: High
Rationale: I would rate the scientific and platform signal as High. This is not merely a phase 1 safety report. It links HSPC engineering, myeloid tumor homing, local IFNα delivery, durable engraftment, low systemic exposure, CSF and tumor IFN activity, single-cell TME reprogramming and TCR clonotype evidence into a relatively coherent early clinical evidence chain.
This High means high scientific and platform signal, not high clinical readiness. Whether Temferon truly improves survival in GBM still requires randomized or at least more persuasive phase 2 data. Conditioning burden and infection risk also need substantial optimization.
I would rate clinical maturity as Medium-Low. The platform has crossed an important first-in-human feasibility and safety threshold and has mechanistic evidence behind it, but the current evidence is still a small, nonrandomized, interim phase 1 dataset. The decisive next test is whether a busulfan-based regimen, recommended phase 2 dose, patient-selection biomarkers and survival endpoints can all hold together.