SEED | 用 prime editing 听见同义突变的沉默信号 SEED | Prime editing hears functional synonymous mutations AI-assisted · reviewed
北京大学、北京大学基因组编辑研究中心和昌平实验室的 Xuran Niu、Wei Tang、Yongshuo Liu 与 Ying Liu、Wensheng Wei 团队近期报道,利用 prime editor 建立 PRESENT 高通量筛选,在人类细胞中系统测试同义突变的适应度效应,发现大多数同义突变接近中性,但少数可通过异常剪接、RNA 折叠和翻译调控产生可测量表型,为临床同义变异解释提供了新的实验与预测框架。
研究问题是什么?
这篇文章问的是一个长期被简化处理的问题:同义突变在人体细胞里到底是不是大多真的“沉默”?
按照遗传密码简并性,同义突变不改变氨基酸序列,因此常被默认接近中性。这个假设在群体遗传学和临床变异注释中都很有影响。但越来越多案例提示,同义突变可能改变 mRNA 剪接、稳定性、结构、翻译速度,甚至与疾病相关。问题在于,人类细胞中经过实验确认的功能性同义突变仍然很少,临床数据库里也有大量同义变异被标为 benign、likely benign 或 VUS。
因此,这篇论文的核心问题不是证明“所有同义突变都有害”,而是更精确地回答三件事:在人类细胞中,同义突变整体上有多大比例会影响细胞适应度;这些少数功能性同义突变通过什么机制起作用;能否把高通量筛选数据转化为临床变异解释工具。
真正的新意是什么?
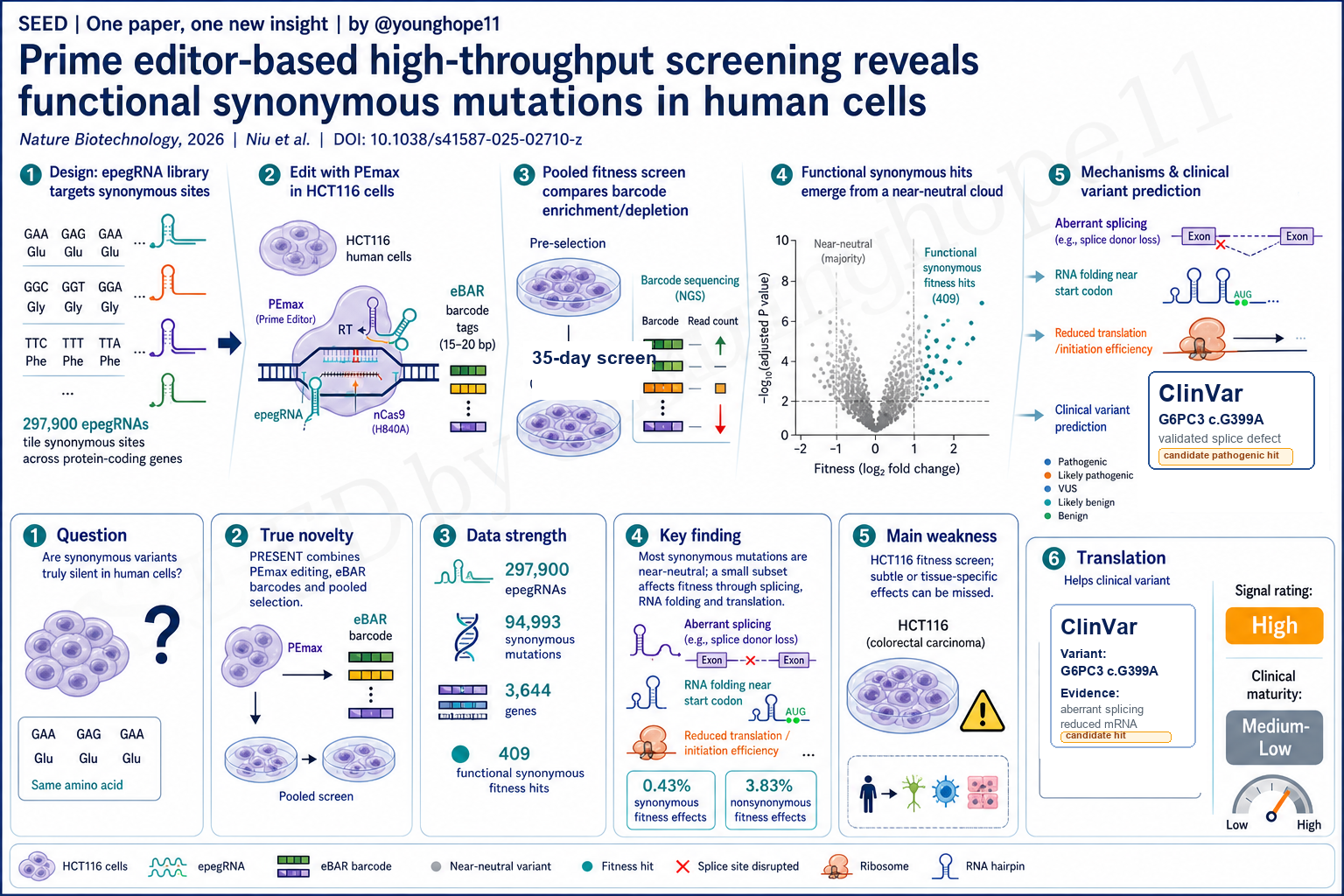
真正的新意是作者把 prime editing 变成了一套同义突变的 pooled functional screen。
他们建立了 PRESENT,也就是 prime editor-based screen technology。这个系统用 PEmax 和 epegRNA 在基因组原位引入单碱基同义突变;每条 epegRNA 外接 eBAR barcode,三个独立 eBAR 相当于内嵌重复;筛选后只需要读出 RTT 和 eBAR,就能从 guide 层面和 mutation 层面估计突变对细胞增殖的影响。
这点很关键。传统 CRISPR knockout 很适合破坏基因,却不适合精确制造大量同义点突变;base editor 受编辑窗口和碱基类型限制;普通 Cas9 切割还可能引入 indel 或结构变异。Prime editor 的优势是可以更精确地写入指定替换,让“同一个氨基酸、不同密码子”的问题被直接测试。
第二个新意是结论本身很克制。作者没有延续酵母研究中“同义突变普遍非中性”的强说法,而是在 HCT116 人类细胞中显示:同义突变和负对照的群体分布更接近,大多数同义突变近似中性;真正值得关注的是少数有明确功能效应的同义突变。
第三个新意是机制和预测工具的连接。作者不仅筛出 409 个影响适应度的同义突变,还开发 DS Finder,把剪接、密码子使用、RNA 折叠、保守性、基因表达和基因 essentiality 等特征整合进模型;又用 DIRECTED-seq 在单细胞水平把 epegRNA、编辑和转录表达联系起来。这使文章从“筛选列表”推进到“机制分类和临床预测”。
数据强在哪里?
数据强在规模、对照和机制验证相互支撑。
筛选库本身很大。作者设计了 297,900 条 epegRNA,覆盖 3,644 个 protein-coding genes,目标包括 94,993 个同义突变和 39,336 个非同义突变。每个突变平均由 2.2 条 epegRNA 覆盖,并包含 AAVS1-targeting 和 nontargeting negative controls。HCT116-PEmax 细胞经过 35 天 pooled screen 后,作者用 ZFC-eBAR 和 RRA 统计框架进行质量控制,设定 RRA <= 0.001,也就是 |screen score| >= 3 的筛选阈值。
在这个阈值下,筛选识别出 1,914 个影响细胞适应度的突变,其中 409 个是同义突变,1,505 个是非同义突变。比例上,只有 0.43% 的同义突变显示可测量适应度效应,而非同义突变为 3.83%。这个数字是论文最重要的信息之一:它同时支持“多数同义突变近中性”和“少数同义突变确实可以有功能效应”。
质量控制也较扎实。作者用 essential genes 上的 nonsense / frameshift perturbations 评估筛选性能,guide 层面 AUC 约为 0.72 或 0.65,mutation 层面约为 0.75 或 0.68,取决于 essentiality 阈值。不同 eBAR 之间在高 LFC 事件中相关性更强,随机选择的高分同义突变也能通过独立编辑和增殖实验验证。作者还专门测试低阈值突变,承认 prime editing efficiency 会影响漏检。
机制层面最强的是剪接证据。DS Finder 和 SpliceAI / AbSplice 分析提示,在 depletion 方向的同义突变中,近四分之一可能影响剪接;其中 donor loss 和 donor gain 是主要模式。BUB1B_R322 破坏 exon 7 / intron 7 交界处 splice donor,导致 intron retention 和异常转录本;EEF2_G332 在 exon 内生成新的 donor site,造成 partial exon excision 和 frameshift。两者都伴随 mRNA 下降和细胞增殖受损。
PLK1_S2 是另一个关键机制例子。这个 AGT > AGC 同义突变位于 PLK1 起始密码子附近,不改变 serine,但预测会增强局部 mRNA 结构稳定性。Western blot 显示 PLK1 蛋白下降;Ribo-seq 显示 start codon 附近 ribosome binding 更困难,而 RNA-seq 没有显示相应转录下降。这支持一个更细的机制:同义突变可以通过改变起始区域 RNA folding 来降低翻译启动效率。
DIRECTED-seq 则把单细胞表达读出接了进来。作者针对 370 个同义突变构建 pooled library,14 天后同时捕获 epegRNA 和转录组,回收 129,193 个 single cells,平均每个细胞 5.5 条 epegRNA,每个目标同义突变平均约 1,692 个细胞覆盖。在 10% FDR 下,他们识别出 40 个显著影响对应基因表达的同义突变。EEF2_G332 使 EEF2 表达降到约 32%,RPL11_V132 和 BUB1B_R322 也显著降低表达,与剪接验证相互吻合。
最后,作者把模型用于 ClinVar。以结肠相关疾病变异为起点,DS Finder 评分 585 个临床记录的同义突变,并把 G6PC3 c.G399A 作为例子验证。这个突变在 ClinVar 中标为 likely benign,但 DS Finder 预测得分最高,实验显示它产生异常转录本并降低 G6PC3 mRNA。模型阈值方面,0.138 阈值在 5% false positive rate 下召回 21/45 个已知 pathogenic synonymous mutations;更严格的 0.370 阈值对应 1% false positive rate,给出 3 个 likely pathogenic candidates。作者还在 A549 和 KYSE-30 中重复 PRESENT 并训练模型,提示细胞背景确实重要。
最大弱点是什么?
最大弱点是筛选主要建立在 HCT116 这一 MMR-deficient 结直肠癌细胞系里。HCT116 对 prime editing 友好,但它不是正常细胞,也不是许多临床疾病最相关的细胞类型。同义突变的影响高度依赖基因表达、剪接环境、组织背景、细胞状态和选择压力;在 HCT116 中近中性,不代表在神经元、造血细胞、肝细胞或发育系统中也一定近中性。
第二个限制是检测灵敏度受 prime editing efficiency 影响。作者很诚实地指出,低编辑效率或弱适应度效应的突变可能被漏掉。严格阈值降低了 false positive,但也会牺牲 subtle effect 和 context-specific effect 的发现能力。因此,0.43% 不能被理解为人类所有同义突变真实功能比例的最终答案,它更像是在这个细胞系统、这个编辑平台和这个统计阈值下的可检测比例。
第三个限制是 readout 以细胞适应度和表达为主。很多同义突变可能不改变短期增殖,却影响 stress response、药物敏感性、分化、蛋白折叠质量控制、免疫识别或特定组织功能。PRESENT 和 DIRECTED-seq 已经提供了平台,但这篇论文还没有系统覆盖这些更复杂的 phenotype space。
第四个限制是 DS Finder 还不是临床级变异解释器。它比 SilVA 的训练集更大,也比泛突变工具 CADD 更贴近同义突变,但模型仍然由特定 pooled screens 训练,且强依赖细胞背景。G6PC3 c.G399A 的验证是在 HCT116 中完成,不是在中性粒细胞发育或患者来源系统中完成。把一个 ClinVar 注释从 likely benign 改到 likely pathogenic,最终仍需要遗传共分离、疾病相关细胞模型、功能 rescue、临床表型和独立队列证据。
最后,文章告诉我们同义突变不是全都沉默,但它没有把所有机制都关上。剪接、RNA folding、codon usage 和表达调控是重要路径,但 RNA-binding proteins、mRNA localization、translation elongation dynamics、co-translational folding 和 proteostasis 仍有很多未解析空间。
是否有临床转化意义?
有,但主要是变异解释和功能基因组学平台意义,不是直接治疗意义。
第一,它提醒临床遗传学不要机械忽略同义突变。多数同义突变可能仍然接近中性,但如果一个同义变异位于 splice donor / acceptor 邻近区域、改变 GC3、影响高表达或 essential gene、改变局部 RNA folding,尤其又出现在相关疾病基因中,就值得进入更严肃的功能评估。
第二,PRESENT 可以作为同义变异功能注释的实验平台。临床数据库里有大量 VUS 和可能误注释的同义变异,仅靠 conservation 或 in silico score 很难判断。Prime editor-based pooled screen 的优势是可以在基因组原位直接写入突变,把变异、细胞适应度、表达变化和机制验证连接起来。
第三,DS Finder 代表一种更实用的方向:不是用一个通用 deleteriousness score 处理所有变异,而是为同义突变这个类别建立专门模型,并允许细胞类型、组织表达和剪接环境进入判断。未来如果在更多疾病相关细胞中扩展,可能会成为 ClinVar reannotation、罕见病变异优先级排序和癌症同义 driver 研究的工具。
但它离临床诊断就绪还有距离。当前数据还不能单独支持临床报告中的 pathogenic / benign 判定,也不能替代 ACMG 框架。更合理的定位是:帮助挑出“值得进一步验证”的同义变异,指导实验设计和证据积累。
Yang 的信号评级:High
理由:我会把这篇论文的科研信号评为 High。它用一个可扩展的 genome editing screen 直接回答了一个长期争论的问题:在人类细胞里,同义突变整体上并不像酵母研究暗示的那样普遍有强适应度效应,但也不是可以被临床和功能基因组学完全忽略的沉默背景。
这个 High 来自三点。第一,实验设计足够大,297,900 条 epegRNA、94,993 个同义突变和 3,644 个基因让结论有群体层面的说服力。第二,作者没有停留在筛选命中,而是通过 BUB1B、EEF2、PLK1 和 G6PC3 等案例把剪接、表达、RNA folding、翻译和临床预测连接起来。第三,PRESENT、DIRECTED-seq 和 DS Finder 组成了一套可以复用的平台,而不是一次性列表。
但这个 High 是“科研和平台信号 High”,不是“临床就绪 High”。临床成熟度我会评为 Medium-Low。原因是主要证据仍来自癌细胞系、pooled fitness readout 和模型预测;真实临床变异解释还需要疾病相关细胞、多组织模型、独立队列和 ACMG 级证据闭环。下一步最重要的是把 PRESENT 扩展到更多 disease-relevant cell types,并测试 DS Finder 预测能否在真实临床问题中稳定提高变异解释质量。
Xuran Niu, Wei Tang, Yongshuo Liu and the team of Ying Liu and Wensheng Wei at Peking University, the Peking University Genome Editing Research Center and Changping Laboratory recently reported PRESENT, a prime editor-based high-throughput screen that systematically tests synonymous variants in human cells and shows that most are near-neutral, while a small subset produces measurable phenotypes through aberrant splicing, RNA folding and translational control, providing a new experimental and predictive framework for clinical synonymous variant interpretation.
What is the research question?
This paper asks a question that has often been simplified: are synonymous variants in human cells truly silent most of the time?
Because of the degeneracy of the genetic code, synonymous variants do not change amino acid sequence and are often assumed to be close to neutral. That assumption matters in both population genetics and clinical variant annotation. Yet a growing number of examples suggest that synonymous variants can alter mRNA splicing, stability, structure, translation speed and disease risk. The problem is that experimentally confirmed functional synonymous variants in human cells remain scarce, while clinical databases contain many synonymous variants annotated as benign, likely benign or VUS.
The central question of this paper is therefore not whether all synonymous variants are harmful. It is more precise: what fraction of synonymous variants has detectable fitness effects in human cells, what mechanisms explain the functional minority, and can high-throughput screening data be converted into a useful framework for clinical variant interpretation?
What is truly new?
The real novelty is that the authors turn prime editing into a pooled functional screen for synonymous variants.
They build PRESENT, or prime editor-based screen technology. The system uses PEmax and epegRNAs to introduce single-base synonymous variants in the genome. Each epegRNA carries an external eBAR barcode, with three independent eBARs serving as embedded replicates. After screening, the RTT and eBAR regions are sufficient to estimate variant effects at both guide and mutation levels.
This matters because standard CRISPR knockout is good at disrupting genes but not at precisely generating large numbers of synonymous point mutations. Base editors are constrained by editing windows and base classes. Cas9 cutting can also introduce indels or structural changes. Prime editing is better suited to directly writing specified substitutions, which makes it possible to test the question of same amino acid, different codon.
The second novelty is the restraint of the conclusion. The authors do not extend the strong claim from yeast that synonymous variants are broadly non-neutral. In HCT116 human cells, synonymous variants behave much more like negative controls at the group level, while nonsynonymous variants show clearer fitness effects. Most synonymous variants are near-neutral; the important signal is the small functional subset.
The third novelty is the connection between mechanism and prediction. The authors do not only identify 409 fitness-affecting synonymous variants. They also develop DS Finder, a model that integrates splicing, codon usage, RNA folding, conservation, gene expression and gene essentiality. They then add DIRECTED-seq, linking epegRNAs, editing and transcript abundance at single-cell scale. This moves the work from a hit list toward mechanism classification and clinical prediction.
Where is the data strongest?
The data are strongest where scale, controls and mechanistic validation reinforce one another.
The screening library is large. The authors design 297,900 epegRNAs across 3,644 protein-coding genes, targeting 94,993 synonymous variants and 39,336 nonsynonymous variants. Each mutation is covered by an average of 2.2 epegRNAs, and the library includes AAVS1-targeting and nontargeting negative controls. After a 35-day pooled screen in HCT116-PEmax cells, the authors process the data with ZFC-eBAR and RRA, using RRA <= 0.001, or |screen score| >= 3, as the screening threshold.
At that threshold, the screen identifies 1,914 mutations affecting cell fitness: 409 synonymous and 1,505 nonsynonymous. In proportion, only 0.43% of synonymous mutations show measurable fitness effects, compared with 3.83% of nonsynonymous mutations. This is one of the paper’s key messages. It supports both sides of the conclusion: most synonymous variants are near-neutral, but a small subset can be functional.
The quality controls are also substantial. The authors use nonsense and frameshift perturbations in essential genes to evaluate screening performance. At the guide level, AUC is about 0.72 or 0.65 depending on the essentiality threshold; at the mutation level, AUC is about 0.75 or 0.68. Correlation among eBARs improves at higher LFC values, and randomly selected high-scoring synonymous variants validate through independent editing and proliferation assays. The authors also test below-threshold mutations and acknowledge that prime-editing efficiency can affect false negatives.
The strongest mechanistic evidence is splicing. DS Finder and SpliceAI / AbSplice suggest that nearly one quarter of depleted synonymous variants may affect splicing, with donor loss and donor gain as major patterns. BUB1B_R322 disrupts the exon 7 / intron 7 splice donor, causing intron retention and abnormal transcripts. EEF2_G332 creates a new donor site within an exon, causing partial exon excision and downstream frameshift. Both cases are accompanied by lower mRNA abundance and impaired cell proliferation.
PLK1_S2 provides a different mechanistic example. This AGT > AGC synonymous change lies near the start codon of PLK1 and does not change serine, but it is predicted to stabilize local mRNA structure. Western blot shows reduced PLK1 protein. Ribo-seq shows more difficult ribosome binding near the start codon, while RNA-seq does not show a matching transcriptional decrease. This supports a more specific mechanism: a synonymous variant can reduce translational initiation by changing RNA folding near the start region.
DIRECTED-seq adds single-cell expression readout. The authors build a pooled library for 370 synonymous variants, capture epegRNAs and transcriptomes after 14 days, and recover 129,193 single cells. Each cell carries an average of 5.5 epegRNAs, and each target synonymous variant is represented by roughly 1,692 cells on average. At 10% FDR, the analysis identifies 40 synonymous variants that significantly affect expression of the corresponding gene. EEF2_G332 reduces EEF2 expression to about 32% of baseline, while RPL11_V132 and BUB1B_R322 also reduce expression, matching the splicing validation.
Finally, the authors apply the model to ClinVar. Starting with colonic disease-related variants, DS Finder scores 585 clinically recorded synonymous variants and highlights G6PC3 c.G399A. ClinVar annotates this variant as likely benign, but DS Finder gives it the highest score; experimental validation shows an abnormal transcript and reduced G6PC3 mRNA. At a threshold of 0.138, DS Finder recalls 21 of 45 known pathogenic synonymous mutations at 5% false positive rate. At the stricter threshold of 0.370, corresponding to 1% false positive rate, it identifies three likely pathogenic candidates. The authors also repeat PRESENT in A549 and KYSE-30 cells and train additional models, reinforcing that cellular context matters.
What is the biggest weakness?
The biggest weakness is that the screen is built mainly in HCT116, an MMR-deficient colorectal cancer cell line. HCT116 is favorable for prime editing, but it is not a normal cell and is not the disease-relevant context for many clinical variants. The effect of a synonymous variant depends heavily on gene expression, splicing environment, tissue context, cell state and selective pressure. Near-neutral in HCT116 does not necessarily mean near-neutral in neurons, hematopoietic cells, hepatocytes or developmental systems.
A second limitation is detection sensitivity. The authors explicitly note that low editing efficiency or weak fitness effects can be missed. A stringent threshold lowers false positives but sacrifices subtle and context-specific effects. The 0.43% estimate should therefore not be read as the final true functional fraction of all human synonymous variants. It is the detectable fraction in this cell system, with this editing platform and this statistical threshold.
A third limitation is the phenotype space. The main readouts are cellular fitness and gene expression. Many synonymous variants may not alter short-term proliferation but could affect stress response, drug sensitivity, differentiation, protein folding quality control, immune recognition or tissue-specific function. PRESENT and DIRECTED-seq provide a platform, but this paper does not systematically cover those more complex phenotypes.
A fourth limitation is that DS Finder is not yet a clinical-grade variant interpreter. It has a larger training base than SilVA and is more tailored to synonymous variants than broad tools such as CADD, but it is still trained on specific pooled screens and remains strongly context-dependent. The G6PC3 c.G399A validation is performed in HCT116, not in neutrophil development or patient-derived systems. Reclassifying a ClinVar variant from likely benign toward likely pathogenic ultimately requires segregation, disease-relevant functional models, rescue, clinical phenotype and independent cohort evidence.
Finally, the paper shows that synonymous variants are not all silent, but it does not close every mechanism. Splicing, RNA folding, codon usage and expression regulation are important routes, but RNA-binding proteins, mRNA localization, translation elongation dynamics, co-translational folding and proteostasis remain open mechanistic space.
Is there translational or clinical relevance?
Yes, but the translational relevance is mainly in variant interpretation and functional genomics infrastructure, not direct therapy.
First, the work warns clinical genetics against mechanically ignoring synonymous variants. Most synonymous variants may still be near-neutral, but if a synonymous variant lies near a splice donor or acceptor, alters GC3, affects a highly expressed or essential gene, changes local RNA folding, and appears in a disease-relevant gene, it deserves more serious functional evaluation.
Second, PRESENT can serve as an experimental platform for synonymous variant annotation. Clinical databases contain many VUS and potentially misannotated synonymous variants, and conservation or in silico scores are often insufficient. A prime editor-based pooled screen can write variants in their genomic context and connect them to fitness, expression and mechanistic validation.
Third, DS Finder points toward a more practical model class: not one universal deleteriousness score for all variant types, but a specialized model for synonymous variants that incorporates cell type, tissue expression and splicing environment. If expanded across more disease-relevant cells, this could support ClinVar reannotation, rare disease variant prioritization and studies of synonymous driver variants in cancer.
But this is not diagnostic-ready. The current data cannot by themselves support clinical pathogenic or benign calls, and they do not replace ACMG-style evidence frameworks. The more appropriate role is prioritization: identifying synonymous variants that deserve deeper validation and guiding the design of follow-up experiments.
Yang’s signal rating: High
Reason: I would rate the scientific signal of this paper as High. It uses a scalable genome editing screen to answer a long-running question directly: in human cells, synonymous variants are not broadly fitness-altering in the way some yeast data suggested, but neither are they a silent background that clinical genetics and functional genomics can ignore.
The High rating comes from three points. First, the experimental design is large enough for a group-level conclusion, with 297,900 epegRNAs, 94,993 synonymous variants and 3,644 genes. Second, the authors do not stop at screening hits; they connect BUB1B, EEF2, PLK1 and G6PC3 examples to splicing, expression, RNA folding, translation and clinical prediction. Third, PRESENT, DIRECTED-seq and DS Finder form a reusable platform rather than a one-off list.
This High means scientific and platform signal High, not clinical readiness High. I would rate clinical maturity as Medium-Low. The main evidence still comes from cancer cell lines, pooled fitness readouts and model prediction; real clinical variant interpretation will require disease-relevant cells, multi-tissue models, independent cohorts and ACMG-grade evidence. The most important next step is to extend PRESENT into more disease-relevant cell types and test whether DS Finder predictions consistently improve variant interpretation in real clinical problems.