SEED | AAV CNS 给药后的 PLAG1 整合警报 SEED | A PLAG1 warning after CNS AAV delivery AI-assisted · reviewed
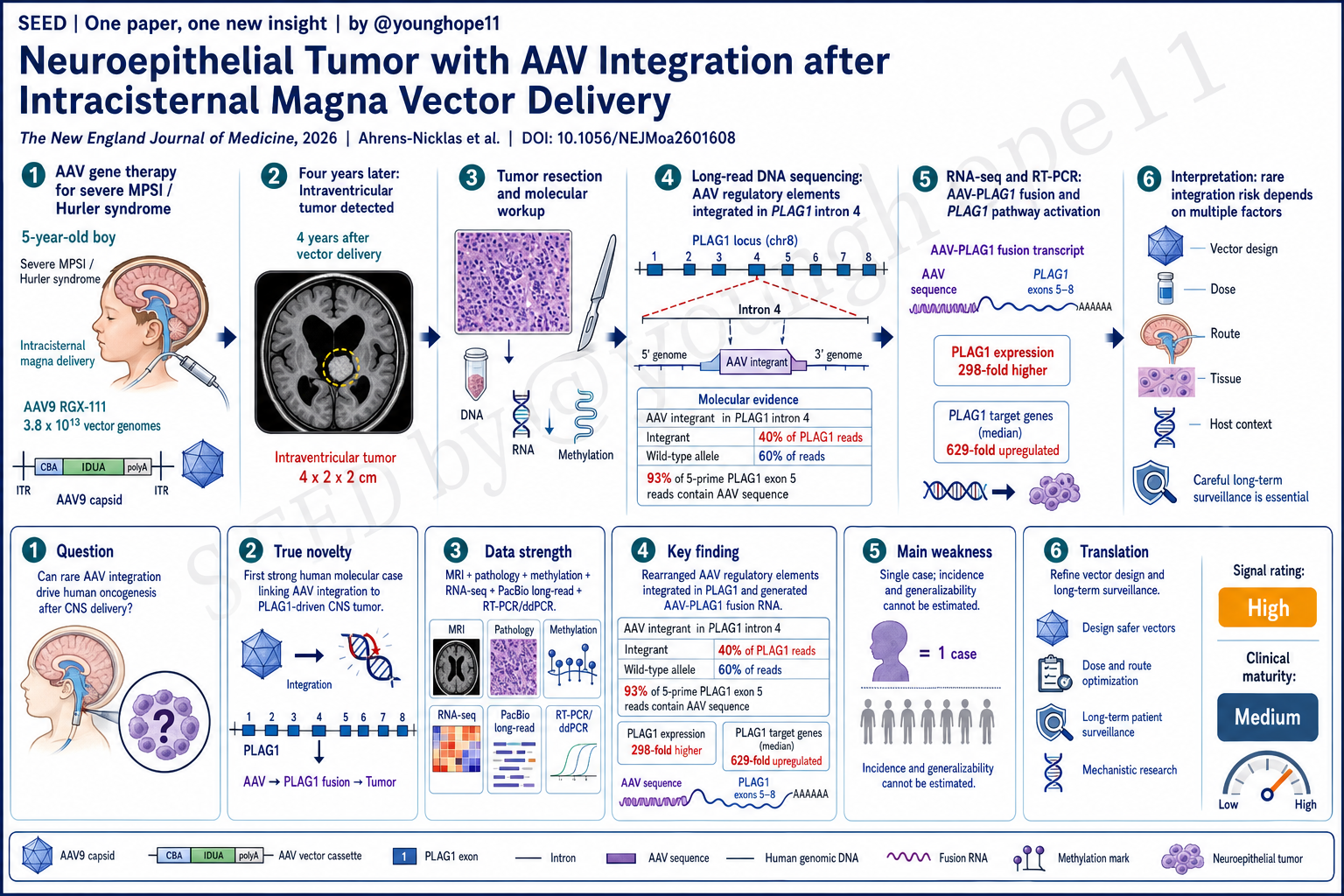
Children’s Hospital of Philadelphia、University of Pennsylvania 和 UC Irvine 的 Rebecca C. Ahrens-Nicklas、Chelsea Kotch 与 Frederic D. Bushman、Lindsey A. George 团队近期报道,一名严重 MPSI/Hurler 综合征男孩在接受枕大池内 AAV9-IDUA 基因治疗 4 年后发生 PLAG1 驱动的神经上皮肿瘤;肿瘤分子分析显示,重排 AAV 调控元件克隆性整合到 PLAG1,并产生 AAV-PLAG1 融合转录本,为 AAV 罕见整合可参与人类肿瘤发生提供了一个强分子证据病例。
研究问题是什么?
这篇 Brief Report 问的是 AAV 基因治疗长期安全性里最敏感的问题之一:AAV 虽然主要以 episomal concatemer 形式存在,但罕见基因组整合是否可能在人类中触发插入性致癌?
过去,AAV integration 的致癌证据主要来自新生小鼠肝癌模型;在犬类中观察过转导细胞的克隆扩增,但尚未有清楚的人类 AAV integration 与 oncogenesis 关联。与此同时,AAV 已经进入多种疾病治疗,作者引用的背景是超过 6000 名患者接受过 AAV gene therapy,整体长期安全性仍然较好。
因此,这篇文章真正要回答的不是“AAV 是否普遍致癌”,而是:在一个具体人类病例中,能不能把 CNS AAV 给药、载体整合、癌基因激活、肿瘤分子类型和临床时间线连接成一条足够强的因果证据链。
真正的新意是什么?
真正的新意是它不是只在肿瘤里“发现 AAV 序列”,而是把 AAV 整合定位到一个有明确肿瘤驱动潜力的基因结构中。
患者是一名严重 MPSI/Hurler 综合征男孩。出生筛查后确诊,4 个月时接受无关供者脐血 HSCT;10 个月时 donor chimerism 降到 27%,13 个月时进入 RGX-111 phase 1-2 临床试验,接受枕大池内 AAV9 载体,总剂量 3.8 x 10^13 vector genomes。该载体包含 CMV enhancer、CBA promoter、CBA intron 和 IDUA transgene,目标是在 CNS 中表达 IDUA。
4 年后,常规脑 MRI 发现 4 x 2 x 2 cm 的脑室内肿块。肿瘤活检组织学不典型,Ki-67 为 5-10%;甲基化提示 PLAGL1-driven tumor,但没有发现 PLAGL1 fusion。RNA-seq 显示 PLAG1 表达比 CHOP CNS tumor cohort 中其他 CNS 肿瘤高 298 倍。最终诊断为 PLAG1-driven neuroepithelial tumor, not elsewhere classified。
最关键的新意在分子结构。RNA-seq 发现 PLAG1 exon 5 与 AAV vector sequence 融合,vector CBA splice donor 接到 PLAG1 exon 5 splice acceptor。PacBio single-molecule long-read DNA sequencing 进一步显示,重排 AAV 片段整合在 chromosome 8 的 PLAG1 intron 4 内,包含 enhancer-promoter-splice donor 结构、IDUA transgene 片段、chromosome 10 片段和 PLAG1 intron 4 短重复。
换句话说,这不是一个随机旁观者事件。整合的 AAV 调控元件方向和结构都支持从强 promoter/enhancer 启动转录,并通过 CBA splice donor 拼接到 PLAG1 exon 5,生成 AAV-PLAG1 融合 RNA。这个融合转录本预计保留 PLAG1 的五个 zinc-finger DNA-binding domains 和 C-terminal transcriptional activation domain,符合 PLAG1 作为 proto-oncogene 被异常激活的机制。
数据强在哪里?
数据强在多层证据彼此闭合,而不是依赖单一检测。
第一层是临床时间线和肿瘤表型。患者在 13 个月时接受 ICM AAV9-IDUA,4 年后 routine neuroimaging 发现脑室内肿瘤;CSF 没有肿瘤播散;肿瘤确认不是 HSCT donor 来源;MRI 和组织病理提示 CNS 肿瘤。患者经过 gross total resection,术后 MRI 发现 3 个不确定意义的亚厘米结节,8 个月随访没有肿瘤增长,认知功能仍高于同龄水平且无神经缺损。
第二层是肿瘤分子分类。甲基化和基因表达支持 PLAG-family tumor。PLAG1 通常主要在胚胎发生中表达,异常激活后可作为 proto-oncogene。这个肿瘤中,PLAG1 exon 5 表达升高,而 PLAG1 exons 1-4 几乎没有表达;在捕获 PLAG1 exon 5 上游序列的 reads 中,93% 含有 AAV sequence,而不是 PLAG1 exon 4,指向 AAV promoter-enhancer 对 PLAG1 exon 5 的选择性上调。
第三层是 DNA 整合证据。PacBio long-read sequencing 在 PLAG1 intron 4 定位到重排 AAV integration structure。这个 integrant 在 reads 中相对丰度为 40%,未受影响 PLAG1 allele 为 60%,与肿瘤起源细胞中一个 PLAG1 等位基因发生 AAV cassette DNA 整合相符。ddPCR 复现了 long-read 定量,targeted PCR 又验证了 integrant 及相关重排结构。
第四层是 RNA 融合验证。肿瘤 RNA-seq 找到 AAV-PLAG1 fusion transcript;两个独立肿瘤 RNA 样本经 RT-PCR 均检测到预期大小产物,而同一患者未受影响脑组织没有该产物;cDNA sequencing 确认了 AAV splice donor 到 PLAG1 exon 5 acceptor 的结构。
第五层是下游 PLAG1 pathway activation。既往 PLAG1 rearranged pediatric CNS tumors 中常上调 IGF2、DLK1、DES、CYP2W1 和 RET;该肿瘤中这 5 个基因全部上调,相比同一患者健康 cortex 的 median fold change 为 629,范围 131 到 13,006。这个下游表达模式支持 AAV-PLAG1 融合不仅存在,而且功能上像 PLAG1-driven tumor。
这些证据合在一起,使这篇文章的强点不是病例数量,而是一个 n=1 病例中罕见但完整的 molecular causality chain:ICM AAV9 exposure -> AAV regulatory element integration into PLAG1 -> AAV-PLAG1 fusion transcript -> PLAG1 target gene activation -> PLAG-family CNS tumor profile。
最大弱点是什么?
最大弱点也是最需要强调的地方:这是一个单病例 Brief Report,不能估算风险发生率,也不能直接外推到所有 AAV 产品、所有给药路径或所有患者。
第二个限制是没有治疗产品库存直接测序验证。作者在 integrant 中发现一个 chromosome 10 fragment,SNP 分析显示它更像来源于 HEK293 producer cells,而不是患者基因组;这提示它可能来自 vector production 过程。但原始 vector product 不可用,因此无法直接在批次产品中确认这个稀有重排结构。即使有库存,超过 10 万亿个给药 vector genomes 中的稀有 packaged variants 也很难被常规深度测序完全捕获。
第三个限制是肿瘤细胞起源和宿主因素仍不完全清楚。ICM AAV9 会在脑室附近产生高局部载体浓度,非人灵长类数据支持 ependymal cells 可被转导,但作者在该患者可用 ependyma 样本中没有观察到这一点。患者本身有 MPSI、HSCT、donor chimerism 下降、免疫调节治疗等复杂背景,是否影响细胞增殖状态或 tumor immunosurveillance,仍无法定量回答。
第四个限制是“关联强”不等于“普遍机制”。该病例显示 AAV integration 可以在特定条件下激活 PLAG1 并参与肿瘤发生,但它没有说明不同 AAV serotype、promoter、dose、route、组织、年龄和疾病状态下风险大小如何变化。作者自己也强调,需要谨慎外推到其他接受 AAV gene therapy 的患者。
最后,检测方法本身也提示未来监测的困难。AAV integrants 常有截短、重排、人-载体嵌合和复杂结构,仅靠 short-read 或单一方法可能漏掉。这个病例之所以有说服力,是因为组织量足够,并且用了 long-read DNA sequencing、targeted PCR、RT-PCR、RNA-seq、methylation profiling 和表达分析。真实世界中很多疑似事件未必有足够组织做这种级别的追踪。
是否有临床转化意义?
有,而且是非常直接的安全转化意义,但它不是在说应该停止 AAV therapy。
第一,它要求 CNS AAV 项目更认真地评估 vector design。强、泛表达 enhancer/promoter 在需要广泛表达的疾病中有治疗合理性,但如果罕见整合发生在 transcription factor 或 proto-oncogene 附近,也可能提供异常启动信号。未来设计需要更系统比较 promoter strength、tissue specificity、self-complementary or single-stranded architecture、ITR/vector backbone fragments 和生产相关重排。
第二,它支持更长时间、更分层的随访。对 CNS delivery,尤其是 ICM/intrathecal 等导致局部高载体浓度的路线,传统短期免疫毒性监测不够。长期 MRI、神经系统评估、CSF 或 tissue biomarker 设计、以及在疑似肿瘤中主动寻找 AAV integration,都会变得更重要。
第三,它提示监管和临床试验设计要把 disease biology 纳入风险评估。MPSI 这类疾病需要早期治疗、CNS 靶向和较强表达,否则无法阻止神经系统进展;风险-收益不能简单套用血友病等低剂量、肝靶向、组织特异 promoter 的经验。真正的问题是为每个疾病和给药路线找到合理的 vector dose、route、promoter 和 surveillance 组合。
第四,它推动检测技术升级。未来 AAV safety workup 不能只问“有没有 vector DNA”,还要问 vector 是否截短、重排、整合到哪里、是否靠近癌基因、是否产生 fusion transcript、是否改变局部基因表达。这个病例说明 long-read sequencing 和 RNA evidence 在判断 genotoxicity 时非常关键。
所以,临床转化意义不是“证明 AAV 不安全”,而是把 AAV 长期安全监测从抽象担忧推进到可检测、可定位、可机制解释的层面。
Yang 的信号评级:High
理由:我会把这篇 Brief Report 的科研和安全信号评为 High。虽然它只有一个病例,但分子证据链非常强:AAV regulatory elements 整合到 PLAG1 intron 4、形成 AAV-PLAG1 fusion RNA、PLAG1 exon 5 选择性上调、PLAG1 target genes 强烈激活、甲基化和表达模式支持 PLAG-family CNS tumor。这不是简单的时间相关性,而是一个结构、转录和肿瘤生物学相互吻合的因果模型。
这个 High 是“罕见但关键的安全信号 High”,不是“发生率 High”。文章不能告诉我们 AAV 肿瘤风险有多高,也不能把一个复杂 MPSI/HSCT/ICM AAV9 病例外推到所有 AAV therapy。它真正改变的是风险框架:AAV integration 在人类中不应只被视为理论风险,至少在特定 vector design、route、dose、组织和宿主背景组合下,可能成为真实 oncogenic event。
临床行动成熟度我会评为 Medium。理由是它已经足够支持更严肃的长期 surveillance、疑似肿瘤的多组学 integration workup、以及对 CNS AAV vector design 的再评估;但还不足以独立决定某类 AAV 治疗是否应停止或大幅改写适应证。下一步最重要的是建立跨产品、跨组织、跨给药路线的长期登记和 integration surveillance,让这个单病例信号转化为可量化的风险分层。
Rebecca C. Ahrens-Nicklas, Chelsea Kotch and the team of Frederic D. Bushman and Lindsey A. George at Children’s Hospital of Philadelphia, the University of Pennsylvania and UC Irvine recently reported a PLAG1-driven neuroepithelial tumor in a boy with severe MPSI/Hurler syndrome 4 years after intracisternal magna AAV9-IDUA gene therapy. Molecular analysis showed clonal integration of rearranged AAV regulatory elements into PLAG1 and expression of an AAV-PLAG1 fusion transcript, providing a strong human molecular case that rare AAV integration can contribute to oncogenesis.
What is the research question?
This Brief Report addresses one of the most sensitive questions in long-term AAV gene therapy safety: although AAV vectors usually persist as episomal concatemers, can rare genomic integration cause insertional oncogenesis in humans?
Before this report, the clearest oncogenic evidence for AAV integration came from neonatal mouse liver cancer models. Clonal expansion of AAV-transduced cells had been observed in dogs, but a clear link between therapeutic AAV integration and human oncogenesis had not been established. At the same time, AAV has become a major therapeutic platform, and the authors note that more than 6000 patients have received AAV gene therapy with an overall favorable long-term safety profile.
The real question is therefore not whether AAV is broadly oncogenic. It is whether, in one human case, CNS AAV exposure, vector integration, oncogene activation, tumor molecular identity and clinical timeline can be connected into a sufficiently strong causal evidence chain.
What is truly new?
The novelty is that the authors do not merely find AAV sequences inside a tumor. They place rearranged AAV regulatory elements inside a gene structure with clear oncogenic potential.
The patient was a boy with severe MPSI/Hurler syndrome. He was diagnosed after newborn screening and received unrelated cord-blood HSCT at 4 months of age. At 10 months, donor chimerism had fallen to 27%. At 13 months, he enrolled in the RGX-111 phase 1-2 trial and received intracisternal magna AAV9 at a total dose of 3.8 x 10^13 vector genomes. The vector contained a CMV enhancer, CBA promoter, CBA intron and IDUA transgene, designed to drive IDUA expression in the CNS.
Four years later, routine brain MRI identified a 4 x 2 x 2 cm intraventricular mass. Tumor biopsy showed indeterminate histologic features with a Ki-67 index of 5-10%. Methylation profiling suggested a PLAGL1-driven tumor, although no PLAGL1 fusion was found. RNA-seq showed PLAG1 expression 298-fold higher than in other CNS tumors from the Children’s Hospital of Philadelphia CNS tumor cohort. The final diagnosis was PLAG1-driven neuroepithelial tumor, not elsewhere classified.
The crucial novelty lies in the molecular structure. RNA-seq found a fusion between PLAG1 exon 5 and AAV vector sequence, with the vector CBA splice donor joined to the PLAG1 exon 5 splice acceptor. PacBio single-molecule long-read DNA sequencing then showed rearranged AAV sequences integrated into PLAG1 intron 4 on chromosome 8, including enhancer-promoter-splice donor elements, IDUA transgene fragments, a chromosome 10 fragment and a short duplication of PLAG1 intron 4.
This is not a random bystander event. The orientation and structure of the integrated AAV regulatory elements support transcription from a strong promoter/enhancer and splicing from the CBA splice donor into PLAG1 exon 5, generating an AAV-PLAG1 fusion RNA. The predicted fusion product retains five PLAG1 zinc-finger DNA-binding domains and the C-terminal transcriptional activation domain, consistent with abnormal activation of PLAG1 as a proto-oncogene.
Where is the data strongest?
The data are strongest because multiple evidence layers close the same loop rather than relying on a single assay.
The first layer is the clinical timeline and tumor phenotype. The patient received ICM AAV9-IDUA at 13 months and developed an intraventricular tumor 4 years later on routine neuroimaging. CSF showed no tumor dissemination. The tumor was confirmed not to originate from the HSCT donor. MRI and histopathology supported a CNS tumor. After gross total resection, MRI showed three subcentimeter nodules of uncertain significance; at 8 months after resection, there was no evidence of tumor growth, and the child had advanced cognitive function for age with no neurologic deficits.
The second layer is tumor classification. Methylation and gene-expression profiling supported a PLAG-family tumor. PLAG1 is normally expressed mainly during embryogenesis, but when dysregulated it can act as a proto-oncogene. In this tumor, PLAG1 exon 5 expression was elevated, while PLAG1 exons 1 through 4 showed little to no expression. Among reads capturing sequence 5 prime of PLAG1 exon 5, 93% contained AAV sequence rather than PLAG1 exon 4, pointing to selective upregulation of PLAG1 exon 5 by the AAV promoter-enhancer.
The third layer is DNA integration evidence. PacBio long-read sequencing identified a rearranged AAV integration structure in PLAG1 intron 4. The integrant represented 40% relative abundance in the reads, while the unaffected PLAG1 allele represented 60%, consistent with integration of AAV cassette DNA into one of two PLAG1 alleles in the tumor cell of origin. ddPCR reproduced the long-read quantification, and targeted PCR verified the integrant and associated rearrangements.
The fourth layer is RNA fusion validation. Tumor RNA-seq detected the AAV-PLAG1 fusion transcript. RT-PCR in two independent tumor RNA samples detected the expected product, while unaffected brain tissue from the same patient did not. cDNA sequencing confirmed the AAV splice donor to PLAG1 exon 5 acceptor structure.
The fifth layer is downstream PLAG1 pathway activation. Prior PLAG1-rearranged pediatric CNS tumors consistently upregulate IGF2, DLK1, DES, CYP2W1 and RET. All five genes were upregulated in this tumor, with a median fold change of 629 and a range from 131 to 13,006 compared with healthy cortex from the same patient. This downstream expression pattern supports not just the presence of an AAV-PLAG1 transcript, but functional behavior consistent with a PLAG1-driven tumor.
Taken together, the strength of this report is not sample size. It is a rare but unusually complete molecular causality chain in one patient: ICM AAV9 exposure -> AAV regulatory element integration into PLAG1 -> AAV-PLAG1 fusion transcript -> PLAG1 target gene activation -> PLAG-family CNS tumor profile.
What is the biggest weakness?
The biggest weakness is also the point that needs the most emphasis: this is a single-case Brief Report. It cannot estimate incidence and cannot be generalized directly to all AAV products, routes or patients.
A second limitation is that the administered vector product was not available for direct sequencing. The integrant contained a chromosome 10 fragment, and SNP analysis suggested that it more closely matched HEK293 producer-cell consensus and common human alleles than the patient’s genotype, pointing to a possible vector-production origin. But without the product lot, this rare rearrangement could not be confirmed directly in the vector stock. Even if product had been available, rare packaged variants among more than 10 trillion administered vector genomes are difficult to capture exhaustively.
A third limitation is that tumor cell origin and host factors remain incompletely resolved. ICM AAV9 creates a high local vector concentration near the ventricles. Nonhuman primate data support ependymal cell transduction, but the authors did not observe this in the available ependymal samples from this patient. The patient also had MPSI, HSCT, declining donor chimerism and immunomodulatory treatment, all of which could have affected cell proliferation state or tumor immunosurveillance, but none can be quantified from this report.
A fourth limitation is that a strong association in this case is not a universal mechanism. The report shows that AAV integration can activate PLAG1 and contribute to tumor development under particular conditions, but it does not define how risk varies across AAV serotype, promoter, dose, route, tissue, age or disease state. The authors themselves caution against over-extrapolating to other patients who receive AAV gene therapy.
Finally, the detection problem is itself a warning. AAV integrants can be truncated, rearranged and human-vector chimeric. Short-read sequencing or any single method may miss them. This case is convincing because there was adequate tissue and the authors used long-read DNA sequencing, targeted PCR, RT-PCR, RNA-seq, methylation profiling and expression analysis. Many real-world suspected events may not have enough tissue for this level of workup.
Is there translational or clinical relevance?
Yes, and the translational relevance is directly about safety, but the message is not that AAV therapy should be stopped.
First, the report demands more serious assessment of vector design in CNS AAV programs. Strong ubiquitous enhancer-promoter elements may be therapeutically rational for diseases requiring broad expression. But if rare integration occurs near a transcription factor or proto-oncogene, the same regulatory strength could become an abnormal initiation signal. Future design needs more systematic comparison of promoter strength, tissue specificity, vector architecture, ITR or backbone fragments and manufacturing-associated rearrangements.
Second, it supports longer and more stratified follow-up. For CNS delivery, especially ICM or intrathecal routes that can create high local vector concentrations, short-term immune-toxicity monitoring is not enough. Long-term MRI, neurologic assessment, CSF or tissue biomarker strategies and active AAV integration analysis in any suspected tumor become more important.
Third, it suggests that regulation and trial design must account for disease biology. MPSI requires early treatment, CNS targeting and sufficient expression to prevent neurologic progression. Its risk-benefit profile cannot simply be borrowed from hemophilia, where lower-dose systemic delivery and tissue-specific promoters may be feasible. The real task is to define the right vector dose, route, promoter and surveillance plan for each disease and delivery context.
Fourth, it pushes safety testing toward better technology. Future AAV workups cannot ask only whether vector DNA is present. They need to ask whether it is truncated, rearranged, where it integrated, whether it lies near oncogenes, whether it creates a fusion transcript and whether it alters local gene expression. This case shows why long-read sequencing and RNA evidence are central for genotoxicity assessment.
The translational meaning is therefore not that AAV is unsafe. It is that long-term AAV safety monitoring has moved from an abstract concern toward something detectable, localizable and mechanistically interpretable.
Yang’s signal rating: High
Reason: I would rate the scientific and safety signal of this Brief Report as High. It is only one case, but the molecular evidence chain is unusually strong: AAV regulatory elements integrated into PLAG1 intron 4, an AAV-PLAG1 fusion RNA was formed, PLAG1 exon 5 was selectively upregulated, PLAG1 target genes were strongly activated, and methylation plus expression profiles supported a PLAG-family CNS tumor. This is not just temporal association. It is a structural, transcriptional and tumor-biologic model that fits together.
This High means rare but critical safety signal High, not incidence High. The paper cannot tell us how frequent AAV-associated tumors are, and it should not be extrapolated from one complex MPSI/HSCT/ICM AAV9 case to every AAV therapy. What it changes is the risk framework: AAV integration in humans should no longer be treated as only a theoretical concern. Under certain combinations of vector design, route, dose, tissue and host context, it may become a real oncogenic event.
I would rate clinical action maturity as Medium. The evidence is already strong enough to support more serious long-term surveillance, multi-omic integration workup of suspected tumors, and re-evaluation of CNS AAV vector design. But it is not enough by itself to stop a class of AAV therapies or broadly rewrite indications. The next step is to build long-term registries and integration surveillance across products, tissues and delivery routes, so this single-case signal can become quantitative risk stratification.