SEED | 跨物种转录组时钟:拆解衰老的模块图谱 SEED | Cross-species transcriptomic clocks map modular ageing AI-assisted · reviewed
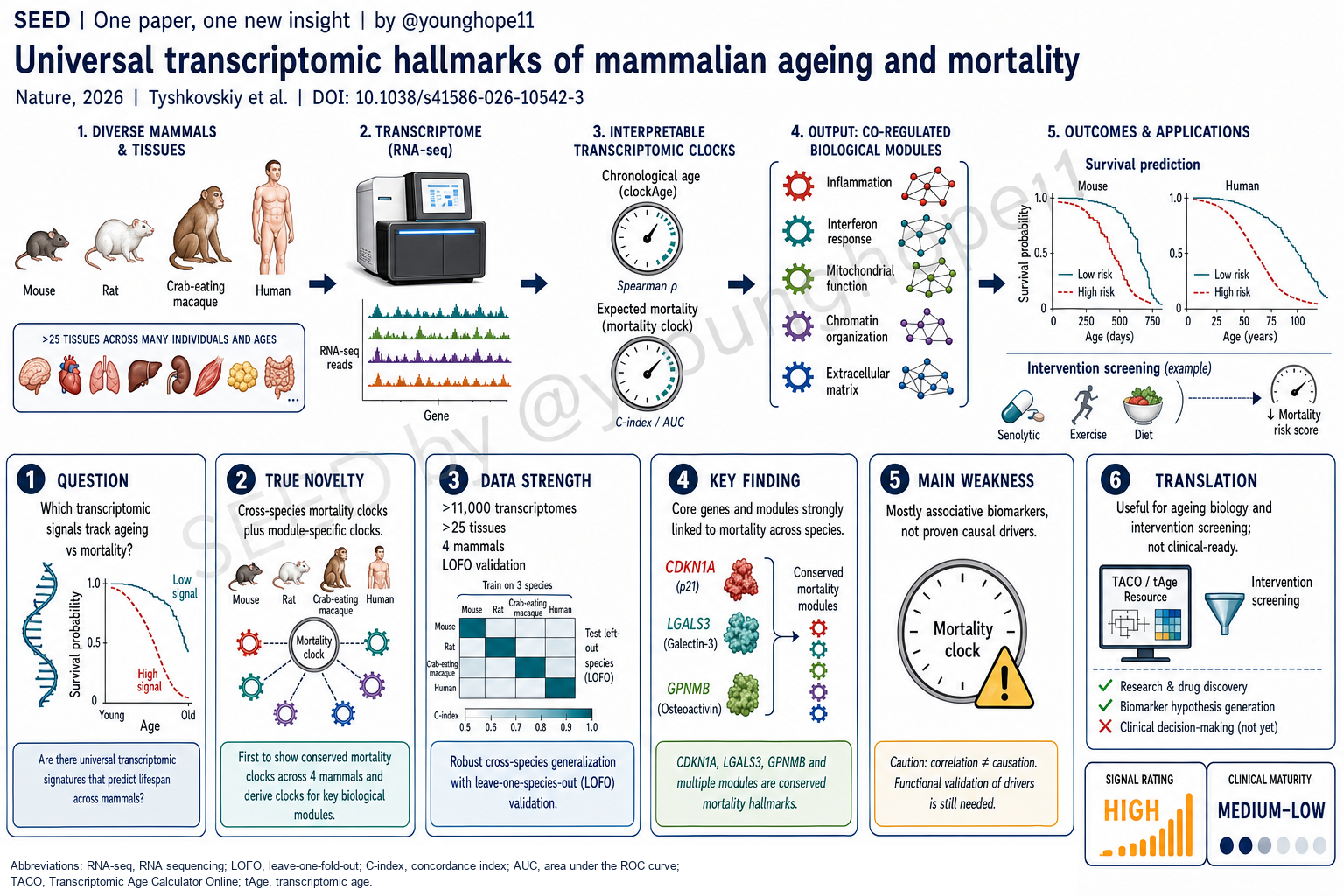
Brigham and Women’s Hospital / Harvard Medical School 的 Alexander Tyshkovskiy 与 Vadim N. Gladyshev 团队近期报道,通过整合来自小鼠、大鼠、食蟹猴和人类的超过 11,000 个、覆盖 25 个以上组织的转录组,建立了可解释的 chronological age 与 expected mortality transcriptomic clocks,并进一步把衰老和死亡风险信号拆解为炎症、干扰素、线粒体、染色质和细胞外基质等模块,为跨物种、跨组织量化细胞子系统的衰老状态提供了新的框架。
研究问题是什么?
这篇文章问的是衰老生物学里一个很核心的问题:在大量随年龄变化的分子信号中,哪些只是时间漂移,哪些更接近死亡风险、疾病负担和寿命干预响应?
传统 biological age clock 多数预测 chronological age。它们能告诉我们样本看起来“更老”还是“更年轻”,但不一定能区分这种变化是否真的贴近健康恶化、寿命缩短或干预效果。尤其在衰老干预研究中,一个干预可能延长寿命,却不明显逆转所有年龄相关表达变化;如果只看 chronological age clock,就可能错过真正贴近生存结局的信号。
作者因此把问题改写成两个层次。第一,能否用跨组织、跨物种转录组同时建立 chronological age 和 expected mortality biomarkers?第二,能否把这些 biomarkers 拆成可解释的功能模块,让我们知道不同疾病或干预主要推动的是炎症模块、线粒体模块、染色质模块,还是其他细胞子系统?
这个问题的重要性在于,转录组比 DNA methylation clock 更容易映射到基因、通路和细胞状态。如果它能同时具备跨物种泛化、死亡结局相关性和模块级可解释性,就不仅是一个“年龄预测器”,而是一个用来读出衰老机制和干预效果的工具。
真正的新意是什么?
真正的新意不是又做了一个 ageing clock,而是把 chronological age、expected mortality、寿命干预、慢性疾病、损伤模型、rejuvenation 模型和模块化通路读数放进同一个转录组框架里。
第一层新意是 expected mortality clock。作者先在 UM-HET3 小鼠中生成 20 种 Interventions Testing Program 药物干预的肝脏 RNA-seq 数据,再整合已发表转录组和生存数据,形成 4,539 个啮齿类样本、26 个组织的 meta-dataset。通过 Gompertz survival model,作者为样本计算 expected log hazard rate 和 normalized age,再训练 chronological age、normalized age 和 expected mortality clocks。
这一步很关键。结果显示,chronological clock 能识别一些有害干预带来的加速衰老,但对寿命延长干预不够敏感;相比之下,normalized-age 和尤其是 mortality clocks 能更好地区分 short-lived 与 long-lived models。也就是说,死亡风险时钟比单纯年龄时钟更贴近“分子健康状态”。
第二层新意是跨物种扩展。作者进一步加入 2,623 个食蟹猴样本和 4,003 个人类样本,训练 multi-species、multi-tissue universal clocks。它们在四个物种内的 chronological age prediction Pearson’s r 均达到至少 0.94,mortality clocks 的 r 均达到至少 0.91;所有测试样本上 chronological tAge 与真实年龄的相关性为 r = 0.952,接近 pan-mammalian DNA methylation clock 的 r = 0.953。
第三层新意是模块化。作者用 co-expression network 把衰老和死亡相关基因组织成模块,再训练 module-specific clocks。保留下来的 23 个 rodent module clocks 和 14 个 multi-species module clocks 让同一个样本不再只有一个“总年龄”,而是有炎症、干扰素、线粒体翻译、氧化磷酸化、染色质修饰、ECM/EMT 等多个子系统读数。
这让文章从“预测”走向“解释”。例如慢性疾病主要加速 inflammatory-module ageing;caloric restriction 主要降低代谢和线粒体相关 mortality tAge;Klotho deficiency 主要影响 mitochondrial、metabolic、NRF2 / proteasome 和 mTOR 相关模块。这样的读数比单一 tAge 数值更接近机制层面的衰老地图。
数据强在哪里?
数据最强在四个层面:规模、跨物种验证、干预/疾病外推,以及人类结局连接。
第一是规模和覆盖面。论文整合超过 11,000 个 transcriptomes,来自 25 个以上组织和 4 个哺乳动物物种:mouse、rat、crab-eating macaque 和 human。啮齿类部分覆盖自然衰老、progeroid models、dwarf models、饮食干预、药物干预、慢性疾病和损伤模型;灵长类部分覆盖 macaque 多组织和人类脑、骨骼肌、皮肤、血液等数据。这种广度让“universal hallmarks”不只是单一组织或单一物种的表达模式。
第二是验证设计较扎实。作者不只做随机 train-test split,还使用 leave-one-dataset-out、leave-one-tissue-out、leave-one-species-out 和 leave-one-fold-out 策略,尽量测试模型对未见数据集、未见组织和未见物种的泛化能力。尤其是在 multi-species clocks 中,leave-one-species-out 仍能在未见物种中捕捉衰老轨迹,说明核心转录组特征有一定跨哺乳动物保守性。
第三是 perturbation 证据丰富。Mortality clocks 在 LPS 急性炎症、caloric restriction、Klotho knockout、replicative senescence、γ-irradiation、metabolic inhibition、cell immortalization、induced reprogramming、heterochronic parabiosis 和 early embryogenesis 等场景中给出方向一致的读数。损伤和慢性病模型通常提高 mortality tAge;immortalization、reprogramming、heterochronic parabiosis 和 early embryogenesis 则减弱或逆转部分 mortality-associated features。
第四是人类数据连接。Framingham Heart Study 中,血液 RNA-seq 样本 n = 3,698,DNA methylation 样本 n = 1,790。Universal transcriptomic mortality clocks 与 time to death 显著相关,表现可接近第二代 DNA methylation clocks,同时保留基因和模块层面的解释性。UK Biobank 中超过 50,000 名参与者的 plasma proteomics 进一步显示,GPNMB、CDKN1A 和 LGALS3 蛋白水平与全因死亡、多种疾病和风险因素正相关。
这些结果让 CDKN1A、LGALS3、GPNMB 等标志物不只是模型里的系数,而是在跨物种转录组、慢性病模型、损伤模型和人群蛋白组中反复出现的 mortality-associated candidates。
最大弱点是什么?
最大弱点是它仍然主要是关联性和预测性框架,而不是因果机制证明。论文题目里的 universal hallmarks 很有吸引力,但这里的 hallmark 首先指跨物种、跨模型反复出现的转录组特征;它们是否是死亡风险的驱动因素,还是组织损伤、炎症、细胞组成变化或疾病负担的下游读数,需要额外的功能实验验证。
第二个弱点是 expected mortality 在动物数据中依赖模型化定义。作者通过生存曲线和 Gompertz meta-model 为样本计算 expected hazard 和 normalized age,这很聪明,也让不同干预可以放在同一框架里比较。但它不是每个被测样本都有个体级、前瞻性死亡结局。这个设计适合发现群体水平的 mortality-associated signatures,但不能直接等同于个体预后模型。
第三个弱点是人类验证仍然集中在可获得数据上。Framingham 的血液数据和 UK Biobank 的 plasma proteomics 很重要,但它们还不是前瞻性干预试验,也不能覆盖所有组织。尤其对于脑、肌肉、肾脏、肝脏等衰老相关组织,人类样本的纵向、结局连接和细胞组成控制仍然有限。
第四个弱点是跨数据集整合不可避免受 batch、平台、细胞组成和 ortholog mapping 影响。作者用 relative expression adjustment、YuGene normalization、matched reference groups、outlier filtering 和多种外部验证来降低这些问题,但模型应用到新实验时仍需要合适的 reference group 和 preprocessing pipeline。换句话说,tAge 不是拿任意 RNA-seq 表达矩阵直接一算就可临床解释的数字。
第五个弱点是 module-specific clocks 的精度中等。单个模块 clock 的预测准确度低于 whole-transcriptome composite clocks;这不削弱它们作为机制读数的价值,但提醒我们不要把每个模块 tAge 都当成高精度生物年龄指标。模块读数更适合比较实验组、解释 perturbation direction,而不是直接给个体做确定性判断。
是否有临床转化意义?
有,但更接近衰老研究、干预筛选和 biomarker hypothesis generation,而不是近期临床诊断工具。
第一,它为衰老干预研究提供了更有用的读数。很多 longevity intervention 并不会简单让所有年龄相关表达变化“变年轻”。Mortality clock 和 module-specific clocks 可以帮助判断一个干预是否真的降低了与死亡风险相关的分子状态,以及它主要作用于哪个细胞子系统。这对筛选药物、比较干预机制和设计 follow-up functional experiments 都有价值。
第二,它让 biological age 的解释性更强。DNA methylation clocks 往往预测力强,但 CpG 位点和机制之间的连接不总是直观。转录组 clock 可以直接落到 CDKN1A、LGALS3、GPNMB、炎症通路、线粒体翻译、染色质修饰等层面,更容易提出机制假设,也更容易和单细胞数据、疾病模型和 perturbation 实验连接。
第三,TACO web app 和 tAge R package 提高了可复用性。对实验室来说,这意味着可以把自己的 RNA-seq 数据放进同一套 preprocessing 和 clock framework 中,估计 composite tAge 和 module-specific tAge,再与对照组或干预组比较。真正有用的地方不是给某个样本贴上“几岁”,而是比较不同条件如何推动或逆转特定 ageing modules。
但临床上还不能直接使用。它目前不应被理解为个体寿命预测工具、疾病诊断工具或抗衰产品评估标准。要走向临床,需要前瞻性 cohort、标准化 sample handling、组织和细胞组成校正、干预前后纵向验证、与临床 endpoint 的独立验证,以及明确的决策场景。
Yang 的信号评级:High
理由:我会把这篇论文的科研信号评为 High。它不是单点 biomarker 论文,而是一个跨物种、跨组织、跨干预的衰老转录组框架;>11,000 个 transcriptomes、四种哺乳动物、死亡风险时钟、人类 time-to-death 连接、UK Biobank 蛋白组验证和模块级 clocks 结合在一起,足以改变我们如何阅读“年龄相关表达变化”。
这个 High 是“科研框架 High”,不是“临床就绪 High”。文章最强的价值在于把衰老信号从单一 age score 拆成 expected mortality 和 cellular subsystem modules,让研究者能够更细地比较疾病、损伤、rejuvenation 和 longevity interventions。
临床成熟度我会评为 Medium-Low。它已经有 Framingham 和 UK Biobank 的人类结局连接,也提供了 TACO / tAge 资源;但它仍主要是研究工具,还缺少标准化、前瞻性、干预响应和临床决策场景验证。下一步最关键的不是再做一个更漂亮的 clock,而是证明这些模块读数能指导机制实验、干预筛选,最终在特定 clinical endpoint 上提供增量价值。
Alexander Tyshkovskiy and the team of Vadim N. Gladyshev at Brigham and Women’s Hospital / Harvard Medical School recently reported an interpretable transcriptomic clock framework built from more than 11,000 transcriptomes across more than 25 tissues in mouse, rat, crab-eating macaque and human. The work develops chronological-age and expected-mortality clocks, then decomposes ageing and mortality-linked signals into modules such as inflammation, interferon signalling, mitochondrial function, chromatin modification and extracellular matrix organization, providing a new framework for measuring ageing of cellular subsystems across species and tissues.
What is the research question?
This paper asks a central question in ageing biology: among the many molecular signals that change with age, which are merely time-associated drift, and which are closer to mortality risk, disease burden and response to lifespan-modulating interventions?
Most biological age clocks predict chronological age. They can tell whether a sample looks molecularly older or younger, but they do not necessarily distinguish whether that shift reflects health deterioration, shortened lifespan or a meaningful intervention effect. In ageing-intervention studies, this distinction matters: an intervention may extend lifespan without globally reversing every age-related expression change, so a chronological-age clock can miss the signal most relevant to survival.
The authors therefore reframe the problem in two layers. First, can cross-tissue and cross-species transcriptomes support biomarkers of both chronological age and expected mortality? Second, can those biomarkers be decomposed into interpretable functional modules, so that a disease or intervention can be read as primarily affecting inflammatory, mitochondrial, chromatin or other cellular subsystems?
The importance is that transcriptomic clocks are mechanistically more readable than many DNA methylation clocks. Gene expression can be mapped directly to genes, pathways and cell states. If the framework generalizes across species and tissues while remaining linked to mortality and interpretable at module level, it becomes more than an age predictor; it becomes a tool for reading mechanisms and intervention responses.
What is truly new?
The real novelty is not simply another ageing clock. It is the placement of chronological age, expected mortality, lifespan-modulating interventions, chronic disease, damage models, rejuvenation models and modular pathway readouts into one transcriptomic framework.
The first novelty is the expected-mortality clock. The authors generated liver RNA-seq data from UM-HET3 mice exposed to 20 Interventions Testing Program treatments, then integrated those data with published transcriptomes and survival information to form a rodent meta-dataset of 4,539 samples across 26 tissues. Using Gompertz survival models, they calculated expected log hazard rate and normalized age for samples, then trained clocks for chronological age, normalized age and expected mortality.
This is the conceptual move. The chronological clock detects some detrimental interventions as accelerated ageing, but it is relatively insensitive to lifespan extension. By contrast, normalized-age clocks and especially mortality clocks distinguish short-lived and long-lived models more robustly. The mortality clock is therefore closer to molecular health status than a simple chronological-age clock.
The second novelty is cross-species expansion. The authors add 2,623 crab-eating macaque samples and 4,003 human samples to train multi-species, multi-tissue universal clocks. Within each of the four species, chronological-age prediction reaches Pearson’s r of at least 0.94, and mortality clocks reach r of at least 0.91. Across all test samples, chronological transcriptomic age correlates with true age at r = 0.952, close to the pan-mammalian DNA methylation clock benchmark of r = 0.953.
The third novelty is modularity. The authors use co-expression network analysis to organize ageing- and mortality-associated genes into modules, then train module-specific clocks. The retained 23 rodent module clocks and 14 multi-species module clocks mean that a sample no longer has only one global age score; it can also have inflammatory, interferon, mitochondrial translation, oxidative phosphorylation, chromatin modification, ECM/EMT and other subsystem scores.
This moves the paper from prediction toward interpretation. Chronic diseases primarily accelerate inflammatory-module ageing; caloric restriction lowers mortality transcriptomic age most strongly in metabolic and mitochondrial modules; Klotho deficiency affects mitochondrial, metabolic, NRF2 / proteasome and mTOR-related modules. These readouts are closer to a mechanistic ageing map than a single transcriptomic-age number.
Where is the data strongest?
The data are strongest across four layers: scale, cross-species validation, perturbation generalization and linkage to human outcomes.
First is scale and coverage. The paper integrates more than 11,000 transcriptomes from more than 25 tissues across four mammalian species: mouse, rat, crab-eating macaque and human. The rodent data include natural ageing, progeroid models, dwarf models, dietary interventions, drug interventions, chronic diseases and damage models. The primate data include macaque multi-tissue profiles and human brain, skeletal muscle, skin and blood. This breadth makes the proposed universal hallmarks more than a single-tissue or single-species pattern.
Second is the validation design. The authors use not only random train-test splits, but also leave-one-dataset-out, leave-one-tissue-out, leave-one-species-out and leave-one-fold-out strategies to test generalization to unseen datasets, tissues and species. In the multi-species clocks, leave-one-species-out analyses still capture ageing trajectories in unseen species, supporting conservation of core transcriptomic ageing features across mammals.
Third is the perturbation evidence. Mortality clocks give directionally coherent readouts across LPS-induced acute inflammation, caloric restriction, Klotho knockout, replicative senescence, gamma irradiation, metabolic inhibition, cell immortalization, induced reprogramming, heterochronic parabiosis and early embryogenesis. Damage and chronic disease models generally increase mortality transcriptomic age, while immortalization, reprogramming, heterochronic parabiosis and early embryogenesis attenuate or reverse parts of the mortality-associated signal.
Fourth is the human connection. In the Framingham Heart Study, the authors analyze blood RNA-seq from 3,698 participants and DNA methylation data from 1,790 participants. Universal transcriptomic mortality clocks are significantly associated with time to death, with performance comparable to second-generation DNA methylation clocks while retaining gene- and module-level interpretability. In UK Biobank plasma proteomics data from more than 50,000 participants, GPNMB, CDKN1A and LGALS3 protein levels are positively associated with all-cause mortality, multiple diseases and risk factors.
Together, these results make CDKN1A, LGALS3, GPNMB and related modules more than model coefficients. They recur across cross-species transcriptomes, chronic disease models, damage models and human proteomics.
What is the biggest weakness?
The biggest weakness is that this remains mainly an associative and predictive framework, not a causal proof of mechanism. The phrase universal hallmarks is attractive, but in this paper the hallmarks are first shared transcriptomic features across species and models. Whether they drive mortality risk, or instead report tissue damage, inflammation, cell-composition shifts or disease burden, requires additional functional experiments.
A second limitation is that expected mortality in the animal data is model-defined. The authors use survival curves and Gompertz meta-models to compute expected hazard and normalized age for samples. This is clever and enables comparisons across interventions, but it is not the same as individual-level prospective mortality outcomes for every assayed sample. The design is well suited for discovering group-level mortality-associated signatures, but it should not be read as a direct individual prognosis model.
A third limitation is that human validation is still limited by available datasets. Framingham blood data and UK Biobank plasma proteomics are important, but they are not prospective intervention trials and they do not cover every tissue. For brain, muscle, kidney, liver and other ageing-relevant tissues, longitudinal human sampling, outcome linkage and cell-composition control remain limited.
A fourth limitation is unavoidable in cross-dataset integration: batch effects, platform differences, cell-composition differences and ortholog mapping all matter. The authors reduce these problems through relative expression adjustment, YuGene normalization, matched reference groups, outlier filtering and multiple external-validation strategies. Still, applying the clocks to a new experiment requires an appropriate reference group and preprocessing pipeline. Transcriptomic age is not a clinical number that can be computed from any RNA-seq matrix without context.
A fifth limitation is the moderate accuracy of module-specific clocks. Individual module clocks are less accurate than whole-transcriptome composite clocks. That does not erase their value as mechanistic readouts, but it means each module score should not be treated as a high-precision biological age metric. Module readouts are better suited for comparing experimental groups and interpreting perturbation direction than for deterministic individual-level judgment.
Is there translational or clinical relevance?
Yes, but the relevance is closer to ageing research, intervention screening and biomarker hypothesis generation than to near-term clinical diagnostics.
First, the framework gives ageing-intervention studies a more useful readout. Many longevity interventions do not simply make every age-associated expression change younger. Mortality clocks and module-specific clocks can help determine whether an intervention lowers molecular states linked to mortality risk, and which cellular subsystem is most affected. That is valuable for drug screening, mechanism comparison and functional follow-up experiments.
Second, it makes biological age more interpretable. DNA methylation clocks can be highly predictive, but the path from CpG sites to mechanism is not always direct. Transcriptomic clocks can point to CDKN1A, LGALS3, GPNMB, inflammatory pathways, mitochondrial translation and chromatin modification, making it easier to generate mechanistic hypotheses and connect clocks to single-cell data, disease models and perturbation experiments.
Third, the TACO web app and tAge R package improve reusability. A laboratory can process its RNA-seq data through the same framework, estimate composite and module-specific transcriptomic age, and compare intervention or disease groups against controls. The most useful output is not an isolated age label for a sample, but a comparison of how different conditions accelerate or reverse specific ageing modules.
But the work should not be used clinically yet. It should not be interpreted as an individual lifespan predictor, diagnostic tool or commercial anti-ageing assessment standard. Clinical translation would require prospective cohorts, standardized sample handling, tissue and cell-composition correction, longitudinal pre/post-intervention validation, independent clinical-endpoint validation and a clearly defined decision context.
Yang’s signal rating: High
Reason: I would rate the scientific signal of this paper as High. This is not a single-biomarker paper; it is a cross-species, cross-tissue, cross-intervention transcriptomic framework for ageing. The combination of more than 11,000 transcriptomes, four mammalian species, mortality clocks, human time-to-death association, UK Biobank proteomic validation and module-specific clocks is strong enough to change how we read age-associated expression changes.
This High means research-framework High, not clinical-readiness High. The strongest value is the separation of ageing signals into expected mortality and cellular subsystem modules, allowing researchers to compare disease, damage, rejuvenation and longevity interventions with more resolution than a single age score.
I would rate clinical maturity as Medium-Low. The work includes human outcome links from Framingham and UK Biobank and provides reusable TACO / tAge resources, but it remains primarily a research tool. It still lacks standardization, prospective validation, intervention-response validation and a clinical decision context. The next step is not merely a more polished clock, but evidence that these module readouts can guide mechanistic experiments, intervention screening and eventually provide incremental value for specific clinical endpoints.