SEED | Exa-cel 儿科数据:疗效很干净,清髓是瓶颈 SEED | Pediatric exa-cel: efficacy is clean, conditioning is the bottleneck AI-assisted · reviewed
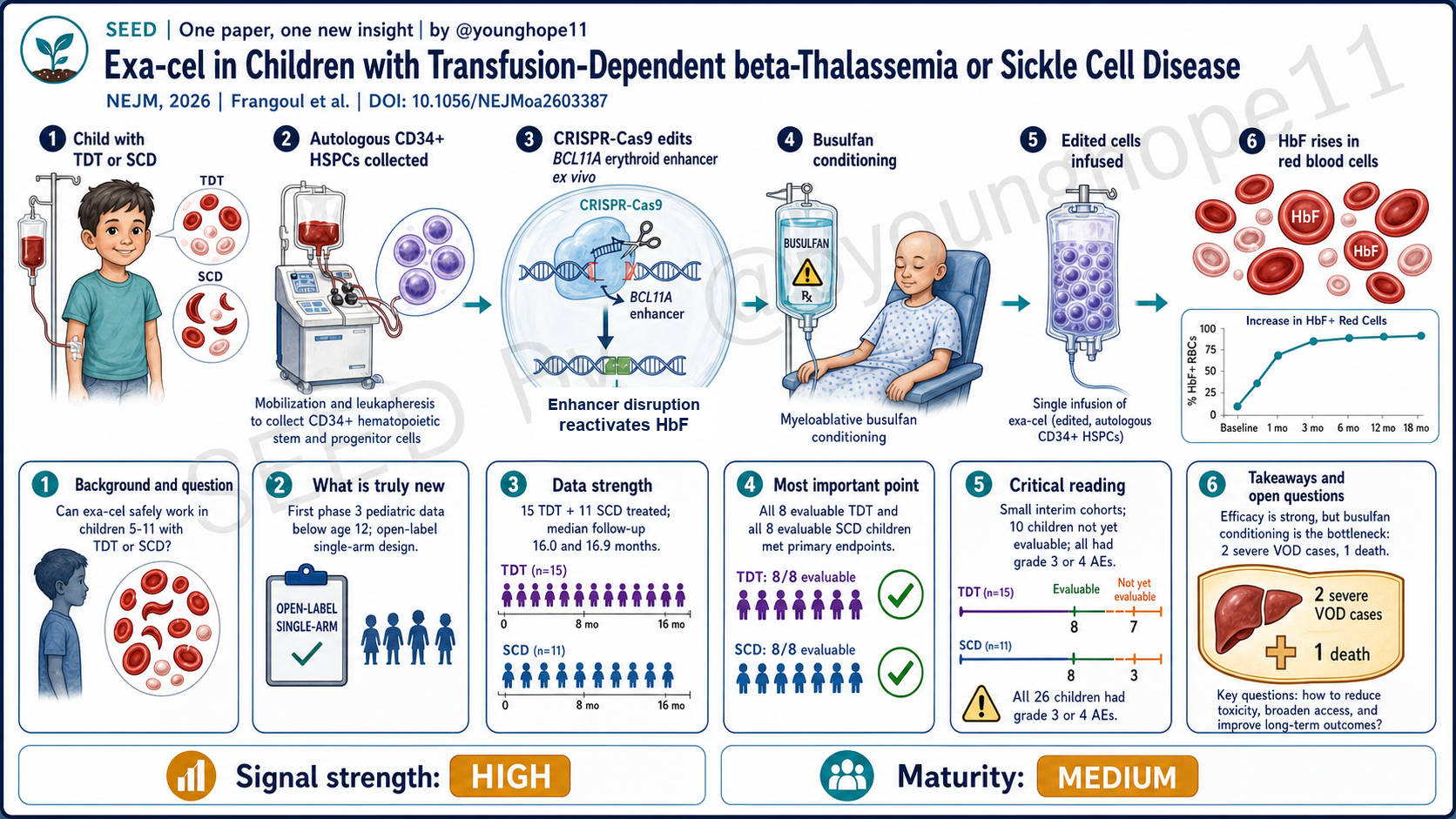
这篇 NEJM Original Article 的真正故事不是“exa-cel 在更小儿童中也有效”,而是:当 BCL11A 编辑已经能稳定制造全细胞分布的 HbF 时,挡在儿童治愈性基因治疗前面的主要瓶颈,正在从 CRISPR 剪刀转移到 busulfan 清髓。 Frangoul、Grupp、Locatelli 与 CLIMB THAL-141 / CLIMB SCD-151 研究组报告了两项 3 期、开放标签、单臂儿科研究的中期结果,把已在 12–35 岁人群建立临床基础的 exa-cel 下探到 5–11 岁儿童。
为什么把 exa-cel 推到 5–11 岁儿童?
输血依赖型 β-地中海贫血(TDT)和镰刀型细胞病(SCD)都是 HBB 相关的严重单基因血红蛋白病。TDT 靠长期输血与铁螯合维持,SCD 则以反复血管闭塞危象、慢性溶血和器官损伤为核心负担。越早在不可逆器官损伤前做治愈性干预,理论获益越大;但适合的同胞供者少于 20%,异基因 HSCT 又带来 GVHD、排斥、免疫抑制和死亡风险。
Exa-cel 的思路不是给一个正常 β-globin,而是取自体 CD34+ 造血干祖细胞,在体外用 CRISPR-Cas9 编辑 BCL11A 的红系特异增强子,降低红系 BCL11A 活性,重新打开 γ-globin / 胎儿血红蛋白(HbF)。此前 12–35 岁 3 期研究中,TDT 输血独立率为 91%,SCD 严重 VOC 消除率为 97%。这篇文章问的是:同一条 BCL11A-HbF 路线能否安全、有效地推到 5–11 岁儿童?
新意不在剪刀,而在儿科风险暴露
这不是新机制论文。BCL11A 增强子编辑、HbF 重激活、exa-cel 在较大年龄段的临床疗效,都已经被前期研究建立。它的新意是三点:
第一,新人群:这是 5–11 岁儿童的 exa-cel 3 期中期数据。儿童不是成人的缩小版,特别是在 busulfan 药代、采集动员、长期生育和远期肿瘤风险上,风险权重完全不同。
第二,硬终点:TDT 的主要终点是连续至少 12 个月输血独立,SCD 的主要终点是连续至少 12 个月无严重 VOC。这些不是替代指标,而是患者真正感受到的疾病负担。
第三,安全性暴露:文章最值得领域反复读的部分不是 8/8,而是 TDT 组 2 例严重肝静脉闭塞病(VOD/SOS)和 1 例死亡,以及之后研究方案把 busulfan 从允许每日一次改为每 6 小时给药。这一处把“清髓毒性”从背景噪音推到前台。
8/8 背后的三条证据链
这组数据强在三个层面同时对上:硬临床终点、血红蛋白机制读数和编辑细胞持久性读数,而不是只靠一个替代指标。
第一,临床终点足够硬。TDT 组有 15 名 5–11 岁儿童接受 exa-cel,中位随访 16.0 个月;其中 8 名随访满 16 个月、进入主要疗效人群的儿童全部达到连续至少 12 个月输血独立,95% CI 为 63–100,平均输血独立持续 23.4 个月。SCD 组有 11 名儿童接受 exa-cel,中位随访 16.9 个月;其中 8 名可评价儿童全部连续至少 12 个月无严重 VOC,95% CI 同样为 63–100,平均无 VOC 持续 19.0 个月。
第二,机制读数和临床终点方向一致。TDT 组第 6 个月总血红蛋白达到 11.9±1.3 g/dL,HbF 为 10.8±1.7 g/dL,并且 ≥99% 红细胞为 F cells。SCD 组第 6 个月总血红蛋白达到 12.2±1.4 g/dL,HbF 为 49.5±6.4%,第 6 个月后 F cells 超过 98%。这说明疗效不是偶然事件,而是与 BCL11A 编辑后 HbF 全细胞分布相吻合。
第三,编辑读数支持植入后的持续性。第 6 个月骨髓 CD34+ 细胞中,BCL11A 等位基因编辑比例在 TDT 组为 77.4±8.5%,在 SCD 组为 84.5±4.9%;外周血编辑比例也在早期建立并保持稳定。这比单纯看到 HbF 上升更有说服力,因为它说明被编辑的造血祖细胞群体确实在体内留下来了。
但强数据也必须和安全性一起读。所有 26 名治疗儿童都有至少一次 3/4 级不良事件;TDT 组出现 2 例严重 VOD/SOS,其中 1 例在第 132 天死亡。这个安全性读数不削弱疗效信号,但决定了“疗效强”和“方案成熟”不能画等号。
两个“别误读”:
- 8/15 不是 53% 应答率。 主要疗效分母只包括随访满 16 个月者;TDT 剩余 7 人、SCD 剩余 3 人是尚未可评价,不是治疗失败。
- 这不是随机对照结论。 两项研究都是开放标签、单臂、描述性中期分析,没有正式样本量计算;95% CI 不能替代假设检验。疗效信号很强,但统计成熟度不能被 100% 这个数字遮住。
真正的瓶颈从编辑转向清髓
这篇文章给领域的非显然信息是:儿科 exa-cel 的核心风险不是编辑本身,而是为植入编辑细胞而必须做的 busulfan 清髓。
疗效端很整齐:两个疾病、两个硬终点、HbF、F cell 全细胞分布、外周血与骨髓编辑率,都朝同一方向走。尤其 SCD 中 HbF 第 6 月约 49.5%,且第 6 月后 F cells 超过 98%,高于 HPFH 共遗传中被认为具有保护意义的约 30% 全细胞 HbF 阈值。这说明 BCL11A 增强子编辑这条因果链在儿童体内仍然成立。
但安全性端更能改变领域判断。所有儿童都有 3/4 级不良事件,TDT 组出现 2 例严重 VOD,其中 1 例死亡。死亡病例第 26 天发生 VOD,继发多器官衰竭和肺炎,第 132 天死亡;研究者和独立监测委员会把总体图景归因为 busulfan 相关 VOD。关键细节是,该患儿接受每日一次 busulfan,累积 AUC 为 93.5 mg·h/L,超过 74–90 的目标范围;事件后方案改成每 6 小时给药,以降低小年龄儿童 busulfan 暴露波动。
这让 exa-cel 的下一阶段问题变得非常清楚:如果疗效已经可以靠 BCL11A-HbF 轴稳定复现,那么真正高杠杆的创新不再是“再把剪刀磨锋利一点”,而是让编辑细胞植入不再依赖基因毒性清髓。对 5–11 岁儿童来说,VOD、不育、远期第二肿瘤和住院移植毒性的权重,比成人更难被“疗效很好”简单抵消。
如何同时读 100% 疗效与 1 例死亡
第一,分母和随访口径要分开看。 全分析集是 TDT 15 例、SCD 11 例;主要疗效人群各 8 例。说“可评价者 100% 达标”准确,说“所有治疗儿童 100% 达标”不准确。
第二,单臂中期数据不能替代长期临床成熟度。 中位随访约 16 个月,只能支持早期疗效与早期毒性判断。编辑后 HSC 克隆是否长期稳定、HbF 是否持久、晚发 MDS/白血病或其他肿瘤风险,都要等 CLIMB-131 的长期随访。
第三,死亡归因值得精读,而不是一句“与 exa-cel 无关”带过。 文中同时写到该死亡病例存在噬血细胞性淋巴组织细胞增多症(HLH),研究者认为可能与 exa-cel 相关;随后作者又根据细胞减少、可溶性 IL-2 受体升高、铁蛋白升高、低 IFN-γ 等总体特征,把诊断落在 busulfan 相关 VOD。这个归因很可能是合理的,但它提醒我们:小儿清髓、炎症、感染和细胞治疗的毒性叙事会叠在一起,不能把复杂病例简化成“编辑零风险”。
第四,外推边界有限。 SCD 研究排除了既往卒中、高卒中风险或 HbF >15% 的儿童;文章只报告 5–11 岁,不覆盖 2–4 岁;研究中心也都具备高级移植与细胞治疗能力。
第五,申办方角色要放在心里。 研究由 Vertex 和 CRISPR Therapeutics 支持,数据收集与分析也由 Vertex 与作者合作完成。这不推翻数据,但会影响我们阅读安全性归因、监管性中期分析和商业化扩展叙事时的警觉度。
下一代治愈方案要绕开清髓
最直接的下一步不是再证明 exa-cel 会升 HbF,而是证明能否在不付出同等清髓代价的情况下保住这份疗效。抗 CD117、抗 CD45 的抗体偶联清髓、其他非基因毒性 conditioning,以及真正绕开采集和清髓的体内 HSC 编辑,都会因为这篇儿科安全性数据而变得更重要。
第二个方向是编辑工具本身的代际替换。本文参考文献 16–18 已把 Cas12a 编辑 HBG1/HBG2 启动子、碱基编辑 HBG 启动子等路线放在坐标系里。Exa-cel 证明了“提高全细胞 HbF 可以改变 TDT/SCD 临床结局”,但不等于 Cas9 双链断裂会长期保持最优。BCL11A 增强子编辑、HBG 启动子编辑、碱基编辑和未来先导编辑,需要在 HbF 分布、编辑率衰减、脱靶、大片段改变和生产复杂度上正面对比。
第三个现实问题是 SCD 的动员采集。SCD 组使用 plerixafor 而不是 G-CSF;仍有 1 名儿童在 3 轮动员和采集后未达到目标细胞剂量而退出。对于小体重儿童,自体采集本身就是平台瓶颈,不是简单的工艺细节。
Yang 的信号评级:High
轴一,信号强度:High。 理由是两个独立疾病、两个硬临床终点和多条机制读数同向:可评价 TDT 儿童 8/8 输血独立,可评价 SCD 儿童 8/8 无严重 VOC,同时总 Hb、HbF、F cell 全细胞分布和编辑率共同支持 BCL11A-HbF 这条因果链。
轴二,临床成熟度:Medium。 不是 Low,因为 exa-cel 在较大年龄段已有强临床基础,这篇儿科数据也有明确机制闭环;但也不是 High,因为样本小、单臂、开放标签、描述性中期分析,10 名治疗儿童主要终点尚不可评价,中位随访只有约 16 个月,并且已经出现 busulfan 相关严重 VOD 和 1 例死亡。
一句话总结:这篇文章把 exa-cel 儿科疗效信号推得更扎实,也把领域真正的下一道门槛照得更清楚:要让儿童基因治疗成为更好的治愈方案,必须先解决清髓,而不只是继续优化剪刀。
The real story in this NEJM Original Article is not simply that “exa-cel also works in younger children.” It is this: once BCL11A editing can reliably generate pancellular fetal hemoglobin, the main bottleneck for curative pediatric gene therapy is shifting from the CRISPR scissors to busulfan conditioning. Frangoul, Grupp, Locatelli and the CLIMB THAL-141 / CLIMB SCD-151 study groups report interim results from two phase 3, open-label, single-arm pediatric studies, moving exa-cel from the older 12–35-year clinical base into children 5–11.
Why move exa-cel into children aged 5–11?
Transfusion-dependent β-thalassemia (TDT) and sickle cell disease (SCD) are severe HBB-related monogenic hemoglobinopathies. TDT is managed with chronic transfusion and iron chelation; SCD is driven by recurrent vaso-occlusive crises, chronic hemolysis and progressive organ injury. A curative intervention earlier in life, before irreversible organ damage, should in principle carry more lifetime value. But fewer than 20% of patients have a suitable HLA-matched sibling donor, and allogeneic HSCT brings graft-versus-host disease, graft rejection, immunosuppression and mortality risk.
Exa-cel does not add a normal β-globin gene. It harvests autologous CD34+ hematopoietic stem and progenitor cells, edits the erythroid-specific enhancer of BCL11A ex vivo with CRISPR-Cas9, reduces erythroid BCL11A activity and reactivates γ-globin / fetal hemoglobin (HbF). Earlier phase 3 studies in ages 12–35 showed 91% transfusion independence in TDT and 97% elimination of severe vaso-occlusive crises in SCD. This paper asks whether the same BCL11A-HbF route can be safely and effectively extended to children 5–11.
The novelty is not the scissors, but pediatric risk exposure
This is not a new-mechanism paper. BCL11A-enhancer editing, HbF reactivation and exa-cel efficacy in older patients were already established. The novelty is in three places.
First, a new population: phase 3 interim data for exa-cel in children 5–11. Children are not smaller adults, especially for busulfan pharmacokinetics, mobilization and collection, fertility risk and late malignancy risk.
Second, hard endpoints: the TDT primary endpoint was transfusion independence for at least 12 consecutive months; the SCD primary endpoint was freedom from severe vaso-occlusive crises for at least 12 consecutive months. These are not merely surrogate biomarkers; they are the disease burdens patients feel.
Third, safety exposure: the most field-relevant part of the paper is not just 8/8. It is the two severe cases of veno-occlusive liver disease (VOD/SOS) in the TDT cohort, one death, and the subsequent protocol amendment changing busulfan from allowing once-daily dosing to every-6-hour dosing. That detail moves conditioning toxicity from background noise to the foreground.
The three evidence chains behind 8/8
The strongest part of the dataset is that three layers line up: hard clinical endpoints, hemoglobin biology and edited-cell persistence, rather than a single surrogate marker.
First, the endpoints are clinically hard. In CLIMB THAL-141, 15 children 5–11 with TDT received exa-cel and median follow-up was 16.0 months; all 8 children who had at least 16 months of follow-up and entered the primary efficacy population were transfusion-independent for at least 12 consecutive months (95% CI, 63–100), with a mean duration of transfusion independence of 23.4 months. In CLIMB SCD-151, 11 children with SCD received exa-cel and median follow-up was 16.9 months; all 8 evaluable children were free of severe VOCs for at least 12 consecutive months (95% CI, 63–100), with a mean VOC-free duration of 19.0 months.
Second, the biology points in the same direction as the clinical endpoints. In TDT, total hemoglobin was 11.9±1.3 g/dL at month 6, HbF was 10.8±1.7 g/dL and at least 99% of red cells were F cells. In SCD, total hemoglobin was 12.2±1.4 g/dL at month 6, HbF was 49.5±6.4% and F cells exceeded 98% after month 6. This makes the result more convincing because the clinical benefit tracks with pancellular HbF induction after BCL11A editing.
Third, the editing readout supports persistence after engraftment. At month 6, edited BCL11A alleles in bone marrow CD34+ cells were 77.4±8.5% in TDT and 84.5±4.9% in SCD, with peripheral-blood editing also established early and then maintained. That is stronger than seeing HbF rise alone, because it suggests the edited hematopoietic progenitor pool persisted in vivo.
The safety data must be read with the same seriousness. All 26 treated children had at least one grade 3 or 4 adverse event. In TDT, two children had severe VOD/SOS and one died on day 132. This does not erase the efficacy signal, but it means “strong efficacy” and “mature regimen” are not the same claim.
Two “do not misread” notes:
- 8/15 is not a 53% response rate. The primary efficacy denominator includes only children with at least 16 months of follow-up; the remaining 7 in TDT and 3 in SCD were not yet evaluable, not treatment failures.
- This is not a randomized controlled conclusion. Both studies are open-label, single-arm, descriptive interim analyses with no formal sample-size calculation. The efficacy signal is strong, but the 100% figure should not be confused with statistical maturity.
The real bottleneck shifts from editing to conditioning
The non-obvious field-level message is: in pediatric exa-cel, the core risk is not the editing step itself, but the busulfan conditioning required to make room for edited cells to engraft.
The efficacy side is internally consistent. Two diseases, two hard endpoints, HbF, pancellular F-cell distribution, peripheral-blood editing and bone marrow editing all point in the same direction. In SCD, HbF was about 49.5% at month 6 and F cells exceeded 98% after month 6, above the approximately 30% pancellular HbF threshold considered protective in hereditary persistence of fetal hemoglobin. That supports the BCL11A-enhancer causal chain in children.
But the safety side changes how the field should read the result. Every child had a grade 3 or 4 adverse event. In the TDT cohort, two children developed severe VOD and one died. The fatal case developed VOD on day 26, followed by multiorgan failure and pneumonia, and died on day 132; the investigators and independent data monitoring committee attributed the overall picture to busulfan-related VOD. The key detail is that this child received once-daily busulfan, with a cumulative AUC of 93.5 mg·h/L, above the 74–90 target range. After the event, both protocols were amended to every-6-hour busulfan dosing to reduce exposure variability in younger children.
That makes the next-stage problem for exa-cel very clear. If efficacy can be reproduced through the BCL11A-HbF axis, the highest-leverage innovation is no longer simply sharpening the scissors. It is enabling engraftment without genotoxic myeloablation. In children 5–11, VOD, infertility, late second malignancy and transplant-hospitalization toxicity carry more weight than they do in adults.
How to read 100% efficacy alongside one death
First, separate denominator from follow-up. The full analysis set was 15 TDT children and 11 SCD children; the primary efficacy population was 8 in each study. “100% among evaluable children” is accurate. “100% of all treated children met the endpoint” is not.
Second, single-arm interim data are not long-term clinical maturity. Median follow-up was about 16 months. Long-term stability of edited HSC clones, durability of HbF and late MDS, leukemia or other malignancy risk remain questions for CLIMB-131 and its long-term follow-up.
Third, the death attribution deserves close reading, not a quick “unrelated to exa-cel” gloss. The paper also notes that the fatal case had hemophagocytic lymphohistiocytosis (HLH), which the investigator considered possibly related to exa-cel. The authors then point to the overall features - cytopenia, elevated soluble IL-2 receptor, elevated ferritin and low IFN-γ - as consistent with busulfan-related VOD. That attribution may be reasonable, but it shows how pediatric conditioning, inflammation, infection and cell-therapy toxicities can overlap. The case should not be simplified into “editing has zero risk.”
Fourth, generalizability is narrow. The SCD study excluded children with prior stroke, high stroke risk or HbF above 15%; the article reports children 5–11, not the younger 2–4 group; and the participating sites were advanced transplant and cell-therapy centers.
Fifth, keep the sponsor role visible. The studies were supported by Vertex and CRISPR Therapeutics, and data collection and analysis were performed by Vertex in collaboration with the authors. That does not invalidate the data, but it should make readers attentive to safety attribution, regulatory interim-analysis framing and commercialization narratives.
The next curative generation has to bypass conditioning
The immediate next step is not to prove again that exa-cel raises HbF. It is to ask whether the same efficacy can be preserved without the same conditioning cost. Anti-CD117 and anti-CD45 antibody-drug conjugate conditioning, other non-genotoxic conditioning strategies and true in vivo HSC editing that bypasses collection and myeloablation all become more important because of this pediatric safety signal.
The second direction is generational replacement of editing tools. References 16–18 in this paper already put Cas12a editing of the HBG1/HBG2 promoters and base editing of HBG promoters into the coordinate system. Exa-cel proves that raising pancellular HbF can change TDT/SCD outcomes, but it does not prove that Cas9 double-strand breaks will remain the optimal implementation. BCL11A-enhancer editing, HBG-promoter editing, base editing and future prime-editing approaches need head-to-head comparison on HbF distribution, editing-rate decay, off-target effects, large genomic alterations and manufacturing complexity.
The third real-world question is SCD mobilization and collection. The SCD cohort used plerixafor rather than G-CSF; one child still withdrew after three mobilization and apheresis cycles failed to reach the target cell dose. In smaller children, autologous collection is a platform bottleneck, not a minor process detail.
Yang’s signal rating: High
Axis 1, signal strength: High. Two independent diseases, two hard clinical endpoints and multiple mechanistic readouts move in the same direction: 8/8 evaluable TDT children were transfusion-independent, 8/8 evaluable SCD children were free of severe VOCs, and total hemoglobin, HbF, pancellular F-cell distribution and editing rates all support the BCL11A-HbF causal chain.
Axis 2, clinical maturity: Medium. Not Low, because exa-cel has a strong clinical base in older patients and this pediatric dataset has a coherent mechanistic loop. But not High, because the study is small, single-arm, open-label and descriptive; 10 treated children were not yet evaluable for the primary endpoint; median follow-up was only about 16 months; and busulfan-related severe VOD and one death have already appeared.
One-sentence summary: this paper makes the pediatric efficacy signal for exa-cel more convincing, but it also makes the next field bottleneck clearer: to make pediatric gene therapy a better curative option, the field must solve conditioning, not merely keep optimizing the scissors.