SEED AI | Germinal:AI 抗体设计真正快在哪 SEED AI | Germinal shows where AI antibody design gets fast AI-assisted · reviewed
Stanford 与 Arc Institute 的 Luis S. Mille-Fragoso、Brian L. Hie、Xiaojing J. Gao 团队近期报道了 Germinal,一个用于 de novo、表位定向抗体设计的生成式管线。这篇文章值得读,不只是因为它用了 AI,而是因为它把 AI 放进了抗体发现价值链的“设计”段,试图解决 CDR 搜索空间巨大、实验筛选慢、表位控制弱这三个长期瓶颈。
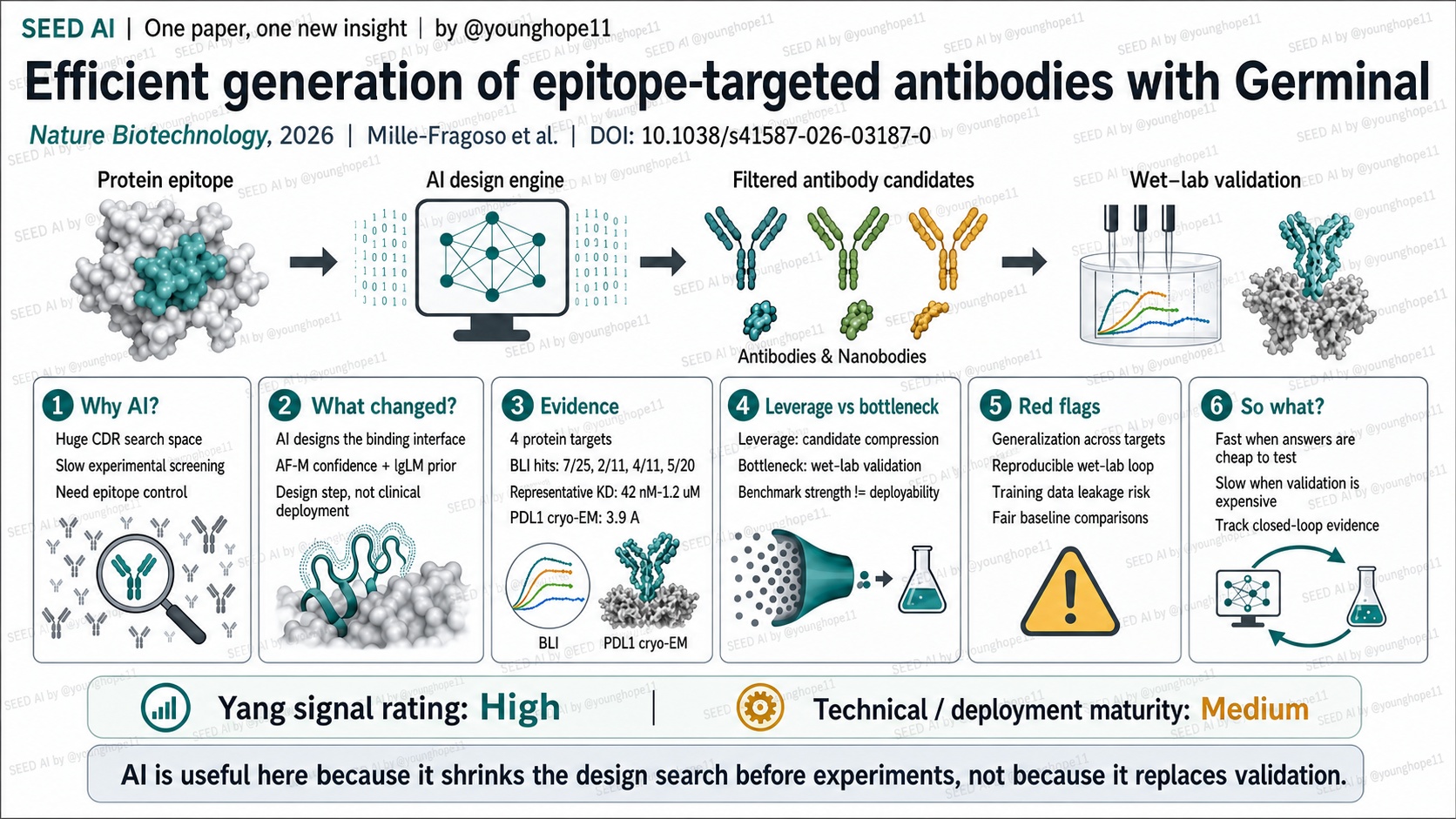
为什么抗体设计适合被 AI 介入
传统抗体发现可以靠动物免疫或大库筛选,但这类流程的核心代价在于实验分母很大,而且很难提前规定抗体必须打到哪个表位。对药物、诊断工具和基础研究试剂来说,“能结合一个蛋白”还不够,很多时候真正重要的是能否结合到功能相关构象或特定表面。
Germinal 对准的不是“再做一个更漂亮的模型”,而是一个很具体的设计瓶颈:在天文级 CDR 序列空间里,提前生成少量更像抗体、也更可能按指定姿势结合目标表位的候选。这里 AI 介入的主因是搜索空间太大,次级瓶颈是实验太慢、预测建模太难;它所在的价值链位置是“设计”,后面紧接湿实验筛选和结构验证。
新意在于把结构可信度和抗体自然性放到同一个优化里
Germinal 改变的是设计到筛选之间的候选生成方式。它把 AlphaFold-Multimer 的结构/复合物可信度和 IgLM 的抗体序列先验合在一起,通过梯度优化 CDR 序列;随后用 AbMPNN 改写非界面 CDR 残基,并用 AF3 作为独立结构预测模型做过滤和排序。
真正的新意不是单纯“用 AI 生成抗体”,而是把两个本来会互相拉扯的目标放进同一个优化问题:结构模型容易推向看起来稳定但不一定像自然抗体的界面,抗体语言模型则约束序列更像真实抗体。作者还加入 paratope- 和 structure-based loss,尽量让结合发生在 CDR 而不是 framework 上,避免模型走向“结构分数好看但生物学姿势不对”的候选。
证据强在四靶点低 n 湿实验闭环
这篇论文最强的证据形态是:四个不同蛋白靶点、每个靶点只测试几十到一百个设计,就在所有靶点上拿到了实验验证的结合分子。纳米抗体部分,作者从 PDL1、IL-3、IL-20 和 BHRF1 分别选择 101、46、43、52 个设计;进入 BLI 的候选分别为 25、11、11、20 个,检测到结合的分别为 7、2、4、5 个。scFv 部分,PDL1 和 IL-3 各测试 48 个设计,SPR 初筛后 BLI 确认 3 个 PDL1 设计和 1 个 IL-3 设计。
验证强度不只是 in-silico 或 benchmark。作者用 split-luciferase、SPR、BLI、polyreactivity assay、cryo-EM 和 alanine mutagenesis 形成了多层闭环。代表性 KD 包括 PDL1 nanobody E11 的 170 nM、IL-3 D2Ser 的 280 nM、IL-20 H5 的 190 nM,以及 BHRF1 C4Lego 的 42 nM;PDL1 scFv H5 还通过 3.9 Å cryo-EM 结构验证,实验结构与预测模型的 global Cα r.m.s.d. 为 1.25 Å。热点突变实验显示,至少一个 alanine substitution 在 17/26 个设计中完全消除了可检测结合,支持其确实打到预测表位。
别误读第一点:benchmark 强不等于落地强。Germinal 的关键不是模型分数本身,而是它把湿实验分母压缩到足够小,让验证闭环变得可负担。别误读第二点:四个靶点全中不等于任意表位都能设计。作者自己也强调,它证明的是可定向设计特定表位,而不是所有表面都可做;hard-to-target surface、非蛋白抗原、糖链、小分子和依赖低质量预测结构的靶点仍在边界之外。
最重要的一点:AI 的杠杆在候选压缩,卡点仍是验证闭环
这篇文章对 AI 生物医药应用边界的核心判断很清楚:AI 在“答案能被便宜验证”的设计任务里已经有真实杠杆。抗体候选是否表达、是否结合、KD 如何、是否打到目标表位,可以通过相对清晰的湿实验和结构方法验证;一旦 AI 把候选数量从成千上万压到几十,实验闭环就从平台级高通量筛选变成普通实验室也可能承担的验证任务。
但它没有绕过验证本身。Germinal 的成功仍依赖 HEK293 表达、BLI/SPR、cryo-EM、突变验证、polyreactivity profiling 和后续蛋白工程修复。真正值得记住的是:这里的 AI 不是替代实验,而是把实验变成更小、更聚焦、更有信息密度的闭环。
批判性阅读:泛化、算力、表位选择和利益绑定
第一,泛化还需要更多靶点和实验室验证。四个蛋白靶点已经比纯 benchmark 强很多,但仍不足以说明它能稳定处理任意膜蛋白、复杂构象、低质量抗原结构、糖链、小分子或非蛋白抗原。作者也承认,Germinal 更适合 favorable protein epitopes,目标表面的生物物理性质会影响成功率。
第二,可复现性比很多 AI 生物论文强,但不是零门槛。代码已在 GitHub 开放,原始 BLI/SPR 数据上传 Zenodo,测试序列和质粒图谱在正文或补充信息中给出;同时,方法部分显示典型采样需要 200-500 H100 GPU hours 才能得到 200-400 个通过过滤的设计,且部分组件如 IgLM、AF3 存在许可或替代实现问题。对普通实验室来说,实验验证分母下降了,但计算和湿实验能力仍是门槛。
第三,数据泄漏和 baseline 要谨慎读。作者做了 PDB/OAS 序列新颖性、结构界面相似性分析,并排除了 AF-M training cutoff 之后的 PDB 结构;这降低了“记住已知复合物”的风险,尤其 IL-3 没有已知抗体或非天然 binder 是重要加分。但这仍不是彻底解决训练集污染问题,只是比常见 AI benchmark 更认真地处理了这个红旗。baseline 方面,文章与 RFDiffusion antibody、mBER、BoltzGen 和行业闭源努力做了讨论,优势主要在低 n、开放方法和结构/突变闭环,而不是宣称全面替代所有抗体发现平台。
第四,利益冲突需要放在读法里。作者中有人是 Arpelos Biosciences、Genyro、Radar Tx 的共同创始人或顾问,且多位作者被列入与本文相关的 Stanford/Arc 临时专利申请。这个不否定结果,但提醒我们:它是一篇强方法论文,也是一条潜在平台资产的公开证明。
对 AI 应用边界的启发
投资或合作判断上,这类平台的价值不只看模型名字,而要看三件事:候选压缩倍率、验证闭环成本、跨靶点泛化。Germinal 给出的信号是,表位定向 antibody design 可能从高通量筛选资产变成设计-验证资产;但平台成熟度仍取决于能否在更多靶点、更多格式、更多实验室里重复低 n 命中。
科研跟踪上,值得继续跟这条线,但要跟的不是单一模型,而是“闭环设计系统”:结构模型、抗体语言模型、过滤指标、湿实验筛选、结构验证和 developability 修复如何协同。接下来最关键的两个问题是:第一,哪些表位性质决定成败?第二,从 binder 到真正 therapeutic antibody,中间需要多少 affinity maturation、format conversion、specificity 和体内验证?
Yang 的信号评级:High
轴一,信号强度:High。理由:这不是单纯 benchmark 论文,而是用四个靶点、低 n 实验分母、BLI/SPR、cryo-EM、热点突变和 polyreactivity profiling 把 AI 设计闭环做了出来。它对“AI 何时能在生物医药落地”给出了很具体的答案:当验证便宜、客观、可闭环时,AI 的候选压缩能力会变成真实生产力。
轴二,技术/落地成熟度:Medium。理由:方法已经接近可被研究型实验室采用,代码和协议也开放;但它仍依赖较高算力、结构预测质量、可选择的蛋白表位和后续湿实验修复。它更像 early discovery compression tool,而不是直接生成临床候选药物的完整平台。
一句话总结:Germinal 的意义不是证明 AI 可以替代抗体实验,而是证明在验证成本足够低的设计任务里,AI 可以把实验从“大海捞针”压缩成几十个候选的闭环判断。
The Stanford and Arc Institute team led by Luis S. Mille-Fragoso, Brian L. Hie and Xiaojing J. Gao recently reported Germinal, a generative pipeline for de novo, epitope-targeted antibody design. The paper matters not simply because it uses AI, but because it places AI in the design step of the antibody discovery value chain, where the long-standing bottlenecks are huge CDR search space, slow experimental screening and weak epitope control.
Why antibody design is a natural AI problem
Traditional antibody discovery can rely on animal immunization or large library screening, but the core cost is the experimental denominator. These workflows often test many candidates and still have limited control over the exact epitope an antibody will recognize. For drugs, diagnostics and basic research reagents, binding a protein is often not enough; the important question is whether the molecule binds a functional conformation or a specific surface.
Germinal is not just asking whether another model can be built. It targets a specific design bottleneck: generating a small set of candidates that look antibody-like and are more likely to bind a user-specified epitope. The primary reason AI enters is that the search space is too large. The secondary bottlenecks are slow experiments and hard prediction. The value-chain position is design, directly upstream of wet-lab screening and structural validation.
The novelty is joint optimization of structural fit and antibody naturalness
Germinal changes the candidate-generation step between design and screening. It combines AlphaFold-Multimer structural or complex confidence with the antibody sequence prior from IgLM, optimizes CDR sequences by gradient-based design, uses AbMPNN to redesign non-interface CDR residues and then uses AF3 as an independent structure model for filtering and ranking.
The real novelty is not simply that AI generates antibodies. It is that Germinal turns two competing objectives into one design problem: structure models can push toward confident but unnatural interfaces, while antibody language models constrain sequences toward realistic antibodies. The authors also add paratope- and structure-based losses to favor CDR-dominated binding rather than framework binding, reducing the risk of designs that score well structurally but use the wrong biological pose.
The evidence is a four-target, low-n wet-lab loop
The strongest evidence is that four distinct protein targets were tested with only dozens to roughly one hundred designs per target, and functional binders emerged for every target. For nanobodies, the authors selected 101, 46, 43 and 52 designs for PDL1, IL-3, IL-20 and BHRF1, respectively. Of those, 25, 11, 11 and 20 advanced to BLI, and detectable binding was observed for 7, 2, 4 and 5 designs. For scFvs, 48 designs each were tested for PDL1 and IL-3; after SPR screening, BLI confirmed 3 PDL1 designs and 1 IL-3 design.
The validation is not only in-silico or benchmark-based. The authors combine split-luciferase screening, SPR, BLI, polyreactivity assays, cryo-EM and alanine mutagenesis into a multi-layer loop. Representative affinities include 170 nM for PDL1 nanobody E11, 280 nM for IL-3 D2Ser, 190 nM for IL-20 H5 and 42 nM for BHRF1 C4Lego. The PDL1 scFv H5 complex was also validated by 3.9-angstrom cryo-EM, with a global C-alpha RMSD of 1.25 angstroms between the experimental structure and the predicted model. In hotspot mutagenesis, at least one alanine substitution abolished detectable binding in 17 of 26 designs, supporting binding to the predicted epitopes.
Do not misread the first point: benchmark strength is not deployability. Germinal’s key contribution is not the model score itself, but the way it makes the wet-lab denominator small enough for validation to close. Do not misread the second point: success across four targets does not mean arbitrary epitopes are solved. The authors themselves emphasize that Germinal demonstrates targeting of specific epitopes, not any arbitrary surface; hard-to-target surfaces, non-protein antigens, glycans, small molecules and targets dependent on poor predicted structures remain outside the demonstrated zone.
The key boundary: AI compresses candidates, but validation still rules
The central boundary judgment is clear: AI has real leverage in design tasks where the answer can be cheaply and objectively validated. Whether an antibody expresses, binds, has a measurable KD and engages the intended epitope can be tested through relatively clear wet-lab and structural assays. Once AI reduces the candidate set from thousands to dozens, validation shifts from a high-throughput platform burden to a focused experimental loop that more labs can plausibly run.
But Germinal does not bypass validation. Its success still depends on HEK293 expression, BLI/SPR, cryo-EM, mutagenesis, polyreactivity profiling and downstream protein-engineering rescue. The important lesson is that AI is not replacing experiments here; it is making experiments smaller, more focused and more information-rich.
Critical reading: generalization, compute, epitope choice and platform incentives
First, generalization still needs more targets and independent labs. Four protein targets are much stronger than a benchmark-only result, but they do not prove that the method can reliably handle arbitrary membrane proteins, complex conformational states, low-quality antigen structures, glycans, small molecules or non-protein antigens. The authors also acknowledge that Germinal is better suited to favorable protein epitopes and that target-surface biophysics will affect success.
Second, reproducibility is stronger than in many AI-biology papers, but it is not frictionless. The code is available on GitHub, raw BLI/SPR data are on Zenodo, and tested sequences plus plasmid maps are provided in the paper or supplementary information. At the same time, the methods report that typical sampling requires 200-500 H100 GPU hours to yield 200-400 designs that pass filters, and some components, including IgLM and AF3, have licensing or open-source substitution issues. The experimental denominator is lower, but compute and wet-lab capacity remain real barriers.
Third, data leakage and baselines deserve careful reading. The authors analyze sequence novelty against PDB and OAS, compare interface structural similarity and exclude PDB structures deposited after the AF-M training cutoff. This reduces the risk that the method merely memorized known complexes, and IL-3 is especially informative because no known antibody or non-natural binder was reported for it aside from its cognate receptor. Still, this does not eliminate training-contamination concerns; it handles the issue more seriously than many AI benchmark papers. On baselines, the paper discusses RFDiffusion antibody, mBER, BoltzGen and industry-led closed-source efforts. Germinal’s main advantage is low-n validation, open methodology and structural/mutational closure, not a claim that it replaces every antibody discovery platform.
Fourth, incentives matter. Some authors disclose outside interests in Arpelos Biosciences, Genyro and Radar Tx, and multiple authors are named on a Stanford/Arc provisional patent application related to the work. This does not invalidate the data, but it should shape the reading: the paper is both a strong methods contribution and a public demonstration of a potential platform asset.
What this means for AI application boundaries
For investment or collaboration judgment, the value of this kind of platform should be judged by three things: candidate compression, validation-loop cost and cross-target generalization. Germinal suggests that epitope-targeted antibody design may shift from a high-throughput screening asset to a design-validation asset. But platform maturity still depends on whether low-n hit rates repeat across more targets, more formats and more independent labs.
For research tracking, this line is worth following, but the object to track is not one model. The important unit is the closed-loop design system: structure model, antibody language model, filtering metrics, wet-lab screen, structural validation and developability repair. The next two questions are clear: which epitope properties predict success, and how much affinity maturation, format conversion, specificity testing and in vivo validation are needed to move from binder to therapeutic antibody?
Yang’s signal rating: High
Signal strength: High. This is not a benchmark-only paper. It builds a design loop across four targets with low experimental denominators, BLI/SPR, cryo-EM, hotspot mutagenesis and polyreactivity profiling. It gives a concrete answer to when AI can matter in biomedicine: when validation is cheap, objective and closeable, candidate compression becomes real productivity.
Technical / deployment maturity: Medium. The method is close to being useful for research labs, and the code plus protocols are public. But it still depends on substantial compute, structure-prediction quality, selectable protein epitopes and downstream wet-lab rescue. It is better understood as an early-discovery compression tool than a complete platform for directly generating clinical antibody candidates.
One-sentence summary: Germinal does not show that AI can replace antibody experiments; it shows that when validation cost is low enough, AI can turn antibody discovery from a large-screen search into a focused loop of dozens of candidates.