SEED | 铁过载重写 HSC 代谢:线粒体失灵后的糖酵解补偿 SEED | Iron overload rewires HSC metabolism toward glycolysis AI-assisted · reviewed
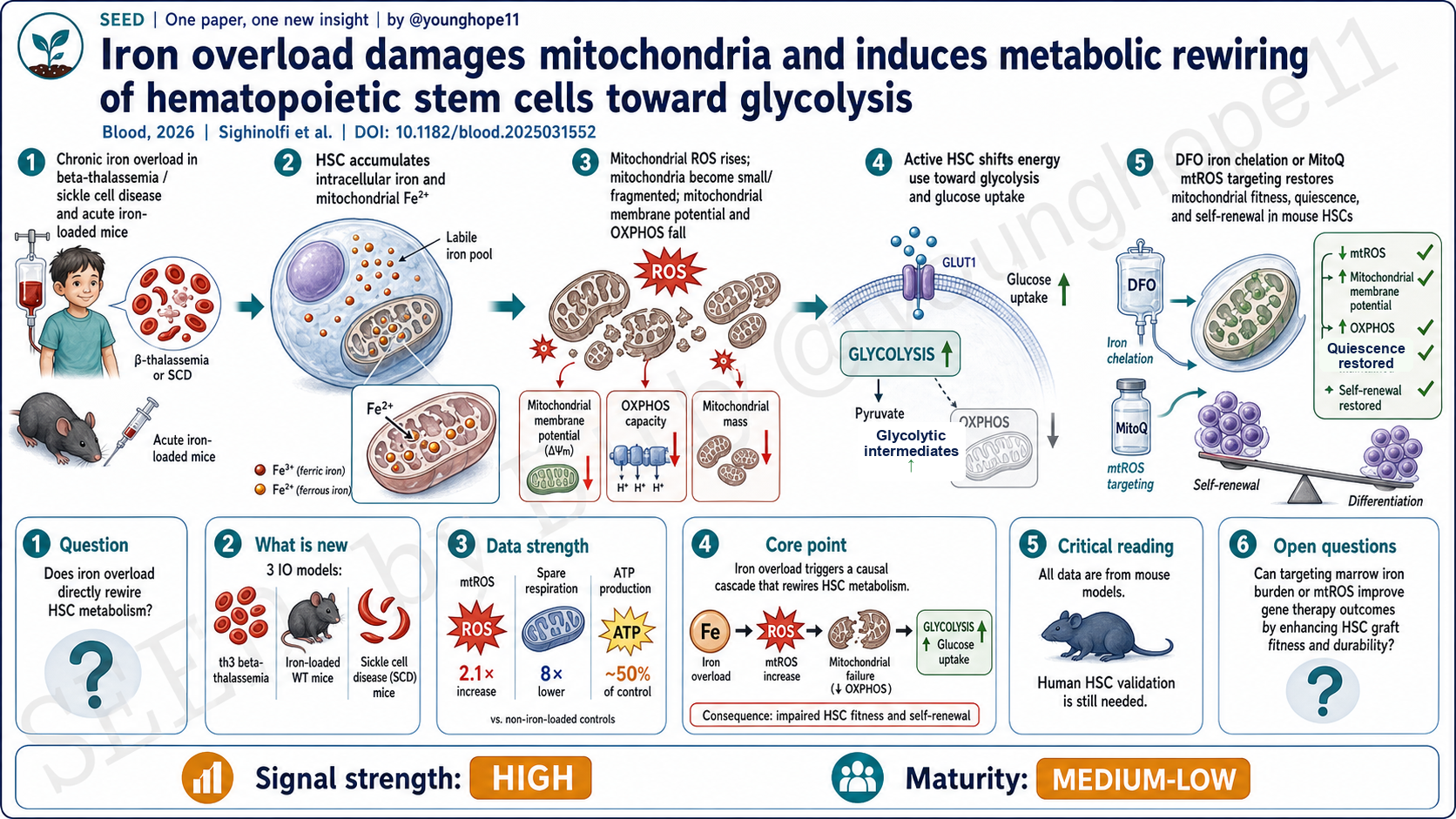
San Raffaele Telethon Institute for Gene Therapy 的 Silvia Sighinolfi、Annamaria Aprile 与 Giuliana Ferrari 团队近期在 Blood 报道,铁过载会在 HSC 中累积为线粒体铁和 mtROS,损伤线粒体膜电位与 OXPHOS,使本来已经被激活的 HSC 转向糖酵解依赖;而体内去铁或靶向 mtROS 可以恢复部分 HSC 静息、自我更新和移植重建能力。
铁过载为什么会成为 HSC 问题?
β-thalassemia 和 sickle cell disease 的铁过载通常被理解为全身器官毒性问题:肝、心、内分泌系统最容易进入临床视野。但对 HSC 来说,问题更隐蔽。HSC 位于骨髓微环境中,既能感受系统性铁负荷,也依赖精细的代谢状态维持静息、自我更新和长期重建能力。
这篇文章问的问题不是“铁多了会不会有毒”这么泛,而是更具体:铁过载是否直接改变 HSC 的线粒体状态和能量代谢,并由此损害 HSC 功能? 为此,作者把疾病模型和干预模型放在同一条因果链上:β-thalassemia 的 th3 小鼠、急性铁负荷 WT 小鼠、SCD 的 SS 小鼠,再加上 DFO 去铁和 MitoQ 降低 mtROS。
新意在于把铁、线粒体和代谢状态连成因果链
已知铁缺乏和铁过载都可能伤害 HSC,也已知 ROS 会推动 HSC 退出静息。但这篇文章的新意是把“铁过载”从背景损伤因素推进到一个更具体的机制轴:iron overload → mitochondrial iron / mtROS → mitochondrial dysfunction / reduced OXPHOS → glycolytic rewiring → HSC exhaustion。
设计上,它不是只用一个 β-thalassemia 模型做描述。作者先在 th3 HSC 中看到铁稳态基因上调、细胞内铁和线粒体 Fe2+ 增加、mtROS 升高、线粒体质量和膜电位下降;再用急性铁负荷 WT 小鼠证明铁本身足以诱导线粒体碎片化、ATP 下降和葡萄糖摄取增加;最后在 SCD 小鼠中验证类似的铁-线粒体-糖酵解重编程。
强数据来自三种模型和两类救援实验
数据最强的地方,是它同时有横向模型复制和纵向功能救援。
在 th3 HSC 中,作者看到线粒体铁增加,mtROS 约 2.1 倍升高,线粒体膜电位下降,电镜显示线粒体变小;功能上,HSC 数量下降,但细胞周期更活跃,G0 静息比例降低。代谢读数也相互吻合:Lin− 祖细胞 Seahorse 显示 spare respiratory capacity 约 8 倍降低,分选 HSC 的总 ATP 约为 WT 的一半,oligomycin 抑制 OXPHOS 后 WT ATP 下降 50%,th3 HSC 只下降 24%,提示 th3 HSC 已经较少依赖 OXPHOS。与此同时,2NBDG 葡萄糖摄取、糖酵解基因和糖酵解中间产物上升。
更关键的是救援实验。DFO 14 天去铁不能纠正 th3 的无效造血或贫血,但能正常化 HSC 的细胞内铁、线粒体铁和 mtROS,恢复 MMP、ATP、葡萄糖摄取、HSC 数量和静息比例,并在二次移植中提高长期重建 HSC 的贡献。MitoQ 5 天不降低铁含量,却降低 mtROS、恢复 MMP 和糖酵解状态,并改善二次移植后的外周血、骨髓和 HSC 嵌合。这说明 mtROS 不是旁观者,而是铁毒性伤害 HSC 功能的重要中介。
真正改变理解的是:活跃 HSC 也会被迫糖酵解
HSC 代谢里常见的简单图景是:静息 HSC 低线粒体活性、偏糖酵解;活化或分化时提高能量需求,并增强 OXPHOS。这篇文章提供了一个更不舒服的状态:铁过载 HSC 是活跃的,但线粒体是坏的。
也就是说,th3 和铁负荷 HSC 并不是健康地从静息进入高能量状态,而是在低 MMP、低 OXPHOS、低 ATP 的背景下被迫增殖。糖酵解在这里不是维持静息的代谢特征,而是线粒体失灵后的补偿方式。这个区分很重要,因为它把 HSC 受损的核心从“ROS 高、细胞更活跃”推进到“铁过载改变了 HSC 能量系统的可用选项”。
对 HSC 移植和基因治疗来说,这一点尤其关键。β-thalassemia 和 SCD 患者常在接受自体 HSC 采集、编辑和回输前已经存在长期铁负荷。如果铁过载让 HSC 更活跃、更依赖糖酵解、更难维持长期自我更新,那么“采到多少 CD34+ 细胞”之外,还要问这些细胞的线粒体和代谢质量是否足够好。
为什么还不能直接外推到患者?
第一,主要证据仍然来自小鼠。th3、SS 和急性铁负荷 WT 模型能拆解机制,但不能等同于人类 β-thalassemia 或 SCD 患者长期输血、螯合治疗和骨髓生态的完整状态。作者也明确指出,仍需要在人类细胞中验证这些结果。
第二,部分代谢读数来自不同层级的细胞群。Seahorse 用的是 Lin− 造血祖细胞,非靶向代谢组用的是 LSK HSPC;作者也做了分选 HSC 的 ATP、2NBDG、MMP 和移植功能读数,但“纯 HSC 内完整代谢通量”仍然很难被直接测量。
第三,DFO 和 MitoQ 是机制工具,不等于现成临床策略。DFO 在 th3 小鼠里改善 HSC 线粒体和移植功能,但并没有纠正无效造血或贫血;MitoQ 改善 mtROS 和 HSC 功能,但人体剂量、骨髓分布、长期安全性和与现有去铁方案的关系,都还不是这篇文章能回答的问题。
第四,骨髓微环境和 HSC 内在效应仍然交织。HSC 的铁、mtROS 和线粒体读数支持细胞内机制,但慢性贫血和铁过载也会改变 niche、炎症和造血压力。未来需要更精细地区分 HSC-autonomous 与 niche-mediated 的部分。
给移植和基因治疗留下的问题
最直接的下一步,是把这条铁-线粒体-代谢轴带到人类 HSC 样本中:不同铁蛋白、肝铁、输血史和螯合状态下,患者 CD34+ / HSC 的线粒体铁、mtROS、MMP、葡萄糖摄取和长期培养/移植替代指标是否真的对应?
第二个方向是治疗窗口。基因治疗和自体 HSC 编辑前,是否应该把“骨髓 HSC 代谢质量”纳入采集前评估?去铁是否需要不只看肝铁和血清铁蛋白,还要看 HSC 或骨髓微环境?mtROS 靶向能否作为采集前短程优化,而不是长期系统治疗?
第三个方向是机制细化。铁为什么进入 HSC 线粒体?是运输、储存、线粒体质量控制、mitophagy 失衡,还是 niche 信号共同驱动?如果能找到更靠近 HSC 的节点,可能比全身去铁或广谱抗氧化更适合转化。
Yang 的信号评级:High
轴一,信号强度:High。 理由是这篇文章不是单一模型描述,而是用 β-thalassemia、急性铁负荷 WT 和 SCD 三个铁过载场景复现同一方向的 HSC 线粒体-代谢改变,并用 DFO 与 MitoQ 两类体内干预把铁、mtROS、线粒体功能、糖酵解和 HSC 自我更新串成了相对完整的因果链。
轴二,成熟度:Medium-Low。 理由是机制证据扎实,但仍主要停留在小鼠和前临床层面;人体 HSC 验证、采集前干预窗口、药物剂量和长期安全性还未建立。
一句话总结:这篇文章把铁过载从“红细胞病的系统性并发症”推进为“会直接重写 HSC 代谢命运的线粒体压力”,也提醒 HSC 基因治疗前的细胞质量评估不能只看数量。
Silvia Sighinolfi, Annamaria Aprile, Giuliana Ferrari and colleagues at the San Raffaele Telethon Institute for Gene Therapy recently reported in Blood that iron overload accumulates as mitochondrial iron and mtROS in HSCs, damages mitochondrial membrane potential and OXPHOS, and pushes already activated HSCs toward glycolytic dependence; in vivo iron chelation or mtROS targeting partially restores HSC quiescence, self-renewal and transplantation fitness.
Why does iron overload become an HSC problem?
In beta-thalassemia and sickle cell disease, iron overload is usually read as a systemic organ-toxicity problem, with liver, heart and endocrine injury dominating clinical attention. For HSCs, the problem is less visible. They sit in the bone marrow microenvironment, sense systemic iron burden, and depend on tightly regulated metabolism to preserve quiescence, self-renewal and long-term repopulation.
The paper does not ask a generic question such as whether too much iron is toxic. It asks something more specific: does iron overload directly change HSC mitochondrial state and energy metabolism, thereby impairing HSC function? To answer that, the authors connect disease models and intervention models along one causal axis: th3 beta-thalassemia mice, acute iron-loaded wild-type mice, SS sickle cell disease mice, plus DFO iron chelation and MitoQ mtROS reduction.
The novelty is linking iron, mitochondria and metabolic state into one causal axis
It was already known that both iron deficiency and iron overload can damage HSCs, and that ROS can push HSCs out of quiescence. What this paper adds is a more concrete mechanism: iron overload -> mitochondrial iron / mtROS -> mitochondrial dysfunction / reduced OXPHOS -> glycolytic rewiring -> HSC exhaustion.
The design is not a single beta-thalassemia model description. The authors first show in th3 HSCs that iron-homeostasis genes are upregulated, intracellular iron and mitochondrial Fe2+ rise, mtROS increases, and mitochondrial mass and membrane potential fall. They then show in acutely iron-loaded wild-type mice that iron itself is sufficient to induce mitochondrial fragmentation, ATP reduction and increased glucose uptake. Finally, they validate a similar iron-mitochondria-glycolysis rewiring in SCD mice.
The strongest data come from three models and two rescue strategies
The strongest part of the dataset is that it combines cross-model replication with functional rescue.
In th3 HSCs, the authors observe increased mitochondrial iron, an approximately 2.1-fold rise in mtROS, lower mitochondrial membrane potential and smaller mitochondria by electron microscopy. Functionally, HSC number decreases, but the cells are more cell-cycle active and less enriched in G0 quiescence. The metabolic data point in the same direction: Seahorse analysis in Lin-negative progenitors shows an approximately eightfold reduction in spare respiratory capacity; total ATP in sorted HSCs is about half that in wild-type controls; and after oligomycin, ATP falls by 50% in wild-type HSCs but only 24% in th3 HSCs, indicating reduced OXPHOS-derived ATP. At the same time, 2NBDG glucose uptake, glycolytic genes and glycolytic intermediates increase.
The rescue experiments are the key. Fourteen days of DFO does not correct ineffective erythropoiesis or anemia in th3 mice, but it normalizes intracellular iron, mitochondrial iron and mtROS in HSCs; restores MMP, ATP, glucose uptake, HSC number and quiescence; and improves the contribution of long-term repopulating HSCs in secondary transplantation. Five days of MitoQ does not lower iron content, but it reduces mtROS, restores MMP and glycolytic state, and improves peripheral-blood, bone marrow and HSC chimerism after secondary transplantation. That makes mtROS a mediator of iron toxicity, not just a bystander.
The important shift is that active HSCs can be forced into glycolysis
A simplified HSC-metabolism model says that quiescent HSCs have low mitochondrial activity and rely more on glycolysis, whereas activated or differentiating HSCs increase energy demand and engage OXPHOS. This paper describes a more uncomfortable state: iron-overloaded HSCs are active, but their mitochondria are damaged.
In other words, th3 and iron-loaded HSCs are not moving healthily from quiescence into a high-energy state. They are proliferating in a context of low MMP, low OXPHOS and low ATP. Here, glycolysis is not simply a quiescence-associated metabolic feature; it is a compensatory route after mitochondrial failure. That distinction matters because it reframes HSC damage from “high ROS and more cycling” to “iron overload changes the energy options available to HSCs.”
For transplantation and gene therapy, this is especially relevant. Patients with beta-thalassemia or SCD may have long-standing iron burden before autologous HSC collection, editing and reinfusion. If iron overload makes HSCs more active, more glycolysis-dependent and less able to maintain long-term self-renewal, then cell quality cannot be judged only by the number of collected CD34+ cells.
Why this should not yet be read as a patient-ready intervention
First, the core evidence is still from mice. The th3, SS and acute iron-loading models are useful for mechanism, but they do not reproduce the full clinical state of human beta-thalassemia or SCD with years of transfusion exposure, chelation history and bone marrow remodeling. The authors explicitly note that human-cell validation is still needed.
Second, some metabolic measurements come from different levels of the hematopoietic hierarchy. Seahorse assays use Lin-negative progenitors, and untargeted metabolomics uses LSK HSPCs. The paper also includes sorted-HSC ATP, 2NBDG, MMP and transplantation assays, but full metabolic-flux measurement inside pure HSCs remains technically difficult.
Third, DFO and MitoQ are mechanistic tools here, not ready-made clinical strategies. DFO improves HSC mitochondrial and transplantation phenotypes in th3 mice, but it does not correct ineffective erythropoiesis or anemia. MitoQ improves mtROS and HSC function, but human dosing, bone marrow distribution, long-term safety and how it would combine with existing chelation regimens are not answered here.
Fourth, the bone marrow niche and HSC-intrinsic effects remain intertwined. The HSC iron, mtROS and mitochondrial data support a cell-intrinsic mechanism, but chronic anemia and iron overload also reshape niche signals, inflammation and hematopoietic stress. Future work needs to separate the HSC-autonomous and niche-mediated components more cleanly.
The next questions for transplantation and gene therapy
The most direct next step is to bring this iron-mitochondria-metabolism axis into human HSC samples. Across ferritin, liver iron, transfusion history and chelation status, do patient CD34+ cells or HSC-enriched fractions show corresponding changes in mitochondrial iron, mtROS, MMP, glucose uptake and long-term functional proxies?
A second direction is the intervention window. Before gene therapy or autologous HSC editing, should marrow HSC metabolic quality become part of pre-collection assessment? Should iron control be judged not only by liver iron and serum ferritin, but also by marrow or HSC state? Could mtROS targeting serve as a short pre-collection optimization strategy rather than a long systemic treatment?
A third direction is mechanism. Why does iron accumulate in HSC mitochondria? Is the driver iron transport, storage, mitochondrial quality control, failed mitophagy, niche signaling, or all of these together? A more HSC-proximal node may be more translatable than broad systemic chelation or generic antioxidant therapy.
Yang’s signal rating: High
Axis 1, signal strength: High. The study is not a single-model description. It reproduces the same HSC mitochondrial-metabolic direction across beta-thalassemia, acute iron loading and SCD, then uses DFO and MitoQ in vivo to connect iron, mtROS, mitochondrial fitness, glycolysis and HSC self-renewal into a relatively coherent causal chain.
Axis 2, maturity: Medium-Low. The mechanistic evidence is strong, but the work remains mainly mouse and preclinical. Human HSC validation, pre-collection intervention windows, dosing and long-term safety are still unresolved.
One-sentence summary: This paper turns iron overload from a systemic complication of hemoglobinopathies into a mitochondrial pressure that rewrites HSC metabolic fate, reminding us that HSC gene therapy should evaluate cell quality, not just cell quantity.